
Integrated Computational Materials Engineering (ICME)
LAMMPS
Overview
The nanoscale material models are molecular dynamics codes and tools used to ascertain properties at the atomistic scale. These simulations generally use interatomic potentials, or force fields, developed using properties obtained from both electronic scale) calculations and experiments, and feed these results into higher scale models, such as dislocation dynamics at the microscale, or continuum models at the macroscale. To date, much of the research at the atomistic scale has focused on informing continuum models for multiscale modeling of metal and polymer material systems. This particular site contains production and research codes that have been developed both at CAVS and outside for performing and analyzing atomistic simulation results. The production codes have user's manuals and a theoretical manual and have been used in practice to solve complex atomistic problems at the nanoscale. The codes that are research codes have not enjoyed the wealth of application and might not have a user's manual or a theoretical manual. We caution the user that there is some risk in using the research version of the codes. Another resource for computational chemistry can be found at computational chemistry.
Finally, to garner more information about the information bridges between length scales go to the Education page.
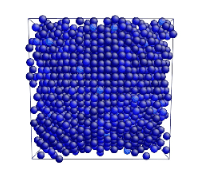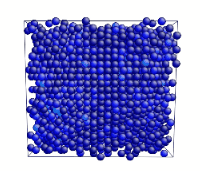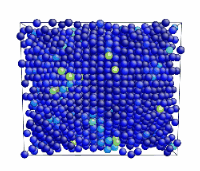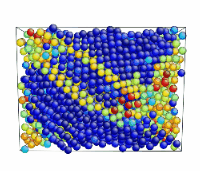
Tensile Loading of an Aluminum Single Crystal. Movie showing deformation of single crystal aluminum loaded in the <100> direction at a strain rate of 1010 s-1 and a temperature of 300 K.
Tutorials
If you are just beginning with atomistic codes, we recommend that you familiarize yourself with LAMMPS, MATLAB (pre- and post-processing), and some of the visualization codes.
LAMMPS
The Basics:- How do I run parallel LAMMPS on LINUX?
- How do I run LAMMPS on a cluster with and without PBS scripting?
- What are the precompiled LAMMPS versions at HPC & CAVS?
- [Installing Linux on Window 10 - Compiling LAMMPS package from the source]
- How to calculate cohesive energy and lattice parameter for aluminum: Part 1
- How to calculate cohesive energy and lattice parameter for aluminum: Part 2
- How to deform a three-dimensional periodic simulation cell in uniaxial tension for aluminum
- How to deform a three-dimensional periodic simulation cell in uniaxial compression for aluminum
- How to generate a Sigma5(310) symmetric tilt grain boundary in aluminum
- How to calculate fracture stress of an iron symmetric tilt grain boundary
- How to deform a nanowire in LAMMPS
- How to construct polymer chains in LAMMPS
- How to construct relaxed bi-layer in LAMMPS
- How to find the generalized stacking fault energy for aluminum
- How to calculate vacancy formation energy for copper
- How to calculate interstitial formation energy for copper
- Reactive Deformation of a Polymer Chain
- Video tutorials for LAMMPS Installation
- Video tutorials for using LAMMPS
- Intro
- Input file Part 1
- Input file Part 2
- Two Element Simulations in LAMMPS Two-element.in.txt AlCu.meam.txt
Visualization
This section shows links to visualization packages used at the atomistic scale. Of these, AtomEye, Ensight, OVITO, and VMD are most frequently used at CAVS. AtomEye, OVITO, and VMD are open source codes.
- AtomEye
- Avizo_(software) - 3d visualization and analysis software.
- Avogadro – Advanced molecule editor and visualizer
- BOSS (molecular mechanics) - MC in OPLS
- Ensight
- iMol – Molecular visualizer for Mac OS X
- JMol – An open-source Java viewer for chemical structures in 3D
- MatLAB - A general purpose technical computing platform with plotting and visualization abilities.
- Molecular Workbench - Interactive molecular dynamics simulations on your desktop.
- OVITO
- Packmol Package for building starting configurations for MD in an automated fashion.
- Punto is a freely available visualisation tool for particle simulations.
- PyMol - Molecular Visualization software written in python.
- RasMol - Molecular Graphics Visualisation Tool
- Sirius visualization software - Molecular modeling, analysis and visualization of MD trajectories.
- Visual molecular dynamics (VMD) - MD simulation trajectories can be visualized and analyzed.
MEAM Parameter Calibration
MEAM Parameter Calibration MPC is a graphical MATLAB application for:
- interactive editing of MEAM library and parameter files,
- running LAMMPS with an input file containing the commands 'pair_style meam' and 'pair_coeff * * LIBRARY_FILE ELEMENTS PARAMETER_FILE ATOM_TYPES', and
- automatic calibration of user-specified MEAM parameters.
See the MEAM Parameter Calibration page.
Tutorial videos for using the MPC tool can be found here, here, and here.
Preprocessing & Postprocessing Codes
This section includes codes used for preprocessing and postprocessing atomistic results. This section can also include scripts used to generate initial structures for inclusion in molecular dynamics simulations. Additionally, this subsection will include examples of xyz coordinate files that can be used in conjunction with the LAMMPS read_data command to upload.
- Initial Structure Generation
- Data Analysis and Plotting
- Visualization
K-12 Projects
This project is designed to help introduce high school students to STEM-related 'relevant' research in physics and materials science and engineering.
Material Models
Molecular Dynamics Codes
This section includes links to molecular dynamics codes. LAMMPS[1] (Large-scale Atomic/Molecular Massively Parallel Simulator) is commonly used for many molecular dynamics simulations related to metal and polymer systems at CAVS. LAMMPS' Fortran predecessor WARP can also be used for parallel molecular dynamics simulations. Last, DYNAMO is commonly used for MEAM (modified embedded atom method)[2] interatomic potential generation.
- LAMMPS Code (see also: LAMMPS tutorials)
- WARP Code
- Gromacs - GROningen MAchine for Chemical Simulation
- IMD - The ITAP Molecular Dynamics program
- NAMD - Not (just) Another Molecular Dynamics program
- OpenMM - OPEN source library for Molecular Modeling simulations
- "Modeling Materials" Other Links
Interatomic Potentials available online
- Interatomic Potentials Repository Project at NIST
- EAM Potential Database at Computational Materials Science Group in Arizona State University
- potfit - the ITAP force-matching Code
For more information on interatomic potential generation using electronic structure information, use the following links.
- The embedded-atom method: a review of theory and applications
- Modified embedded-atom potentials for cubic materials and impurities
- Second nearest-neighbor modified embedded-atom-method potential
- The modified embedded-atom method interatomic potentials and recent progress in atomistic simulations
- Interatomic potentials of the binary transition metal systems and some applications in materials physics
- Parametrization of modified embedded-atom-method potentials for Rh, Pd, Ir, and Pt based on density functional theory calculations, with applications to surface properties
- Density functional theory (DFT)-based modified embedded atom method potentials: Bridging the gap between nanoscale theoretical simulations and DFT calculations
Atomistic Research
This section includes interatomic potential information for atomistic simulations. Embedded atom method[3] potentials can be found at the NIST Interatomic Potential website. A number of modified embedded atom method[2] potentials have been developed here at CAVS for lightweight metals and steel research. Some published and ongoing interatomic potential work at CAVS includes the following:
Metals
- Modified Embedded Atom Method (MEAM) potential for Al-Mg
- MEAM potential for Al, Si, Mg, Cu, and Fe alloys (see also: routines to reproduce the results)
- Grain Boundary Generation of Aluminum[4][5]
- Dislocation Nucleation in Single Crystal Aluminum[6][7][8]
- Uniaxial Tension in Single Crystal AluminumM[8]
- Uniaxial Compression in Single Crystal Aluminum[8]
- Grain Boundary Generation of Copper[4][5]
- Multiscale study of dynamic void collapse in single crystals[9]
- Modified Embedded Atom Method (MEAM) potential for Mg-Al
- Grain boundary generation in Mg[4][5]
- Fatigue Crack Growth Simulation[10]
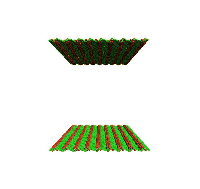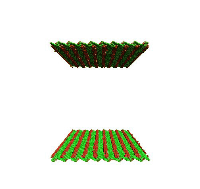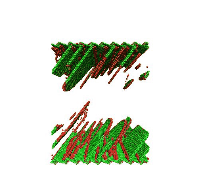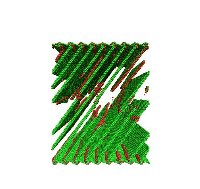
Movie showing dislocation nucleation from a Sigma 3 asymmetric tilt grain boundary
Polymers
Polyethylene
Coarse Grain Simulations
An example of tensile deformation in amorphous polyethylene using a united atom method potential.
All Atom Simulations
Reactive molecular dynamics simulation of hydrocarbon-based polymers, such as polyethylene and polypropylene, is now possible using the recently parameterized modified embedded-atom method (MEAM) potential for hydrocarbons (C/H system).[14] For more information about the potential, please visit the following link:
Polymer Atomistic Research. Movie showing deformation of an amorphous polyethylene structure with 20 chains of 1000 monomers length. The strain rate is 1010 s-1 and the temperature is 100 K[12][13].
Python Based Testing of Atomistic Potentials
- The universal interface for testing atomistic potentials
- Routines for basic tests of atomistic potentials with universal interface
Atomistic Measures of the Elastic and Plastic Deformation Gradient
References
- ↑ S. Plimpton, "Fast Parallel Algorithms for Short-Range Molecular Dynamics," J. Comp. Phys., 117, 1-19 (1995).
- ↑ Baskes, M.I. (1992). Modified embedded-atom potentials for cubic materials and impurities. Phys. Rev. B, 46, 2727 (https://link.aps.org/doi/10.1103/PhysRevB.46.2727).
- ↑ Murray S. Daw, Stephen M. Foiles, Michael I. Baskes,(1993) The embedded-atom method: a review of theory and applications, Materials Science Reports, Volume 9, Issues 7-8, Pages 251-310. (https://dx.doi.org/10.1016/0920-2307(93)90001-U).
- ↑ Tschopp, M. A., & McDowell, D.L. (2007). Structures and energies of Sigma3 asymmetric tilt grain boundaries in Cu and Al. Philosophical Magazine, 87, 3147-3173 (https://dx.doi.org/10.1080/14786430701455321).
- ↑ Tschopp, M. A., & McDowell, D.L. (2007). Asymmetric tilt grain boundary structure and energy in copper and aluminum. Philosophical Magazine, 87, 3871-3892 (https://dx.doi.org/10.1016/j.commatsci.2010.02.003).
- ↑ Spearot, D.E., Tschopp, M.A., Jacob, K.I., McDowell, D.L., "Tensile strength of <100> and <110> tilt bicrystal copper interfaces," Acta Materialia 55 (2007) p.705-714 (https://dx.doi.org/10.1016/j.actamat.2006.08.060).
- ↑ Tschopp, M.A., Spearot, D.E., McDowell, D.L., "Atomistic simulations of homogeneous dislocation nucleation in single crystal copper," Modelling and Simulation in Materials Science and Engineering 15 (2007) 693-709 (https://dx.doi.org/10.1088/0965-0393/15/7/001).
- ↑ Tschopp, M.A., McDowell, D.L., "Influence of single crystal orientation on homogeneous dislocation nucleation under uniaxial loading," Journal of Mechanics and Physics of Solids 56 (2008) 1806-1830. (https://dx.doi.org/10.1016/j.jmps.2007.11.012).
- ↑ K. Solanki, M.F. Horstemeyer, M. I. Baskes, and H. Feng, Multiscale study of dynamic void collapse in single crystals, Mechanics of Materials Volume 37, Issues 2-3, February-March 2005, Pages 317-330 dx.doi.org/10.1016/j.mechmat.2003.08.014
- ↑ Tang, T., Kim, S., & Horstemeyer, M. (2010). Fatigue Crack Growth in Magnesium Single Crystals under Cyclic Loading: Molecular Dynamics Simulation. Computational Materials Science, 48, 426., 48, 426-439 (https://dx.doi.org/10.1080/14786430701255895).
- ↑ Barrett, C.D., El Kadiri, H., Tschopp, M.A. (2011). Breakdown of the Schmid Law in Homogenous and Heterogenous Nucleation Events of Slip and Twinning in Magnesium. Journal of Mechanics and Physics of Solids, in review.
- ↑ Hossain, D., Tschopp, M.A., Ward, D.K., Bouvard, J.L., Wang, P., Horstemeyer, M.F., "Molecular dynamics simulations of deformation mechanisms of amorphous polyethylene," Polymer, 51 (2010) 6071-6083.
- ↑ Tschopp, M.A., Ward, D.K., Bouvard, J.L., Horstemeyer, M.F., "Atomic Scale Deformation Mechanisms of Amorphous Polyethylene under Tensile Loading," TMS 2011 Conference Proceedings, accepted.
- ↑ S Nouranian, MA Tschopp, SR Gwaltney, MI Baskes, and MF Horstemeyer, An Interatomic Potential for Saturated Hydrocarbons Based on the Modified Embedded-Atom Method, Physical Chemistry Chemical Physics 16 (13)(2014):6233-6249.