
Integrated Computational Materials Engineering (ICME)
LAMMPS Interstitial Fault Energy
Abstract
This example shows how to calculate interstitial formation energy (IFE) for FCC metals. The parallel molecular dynamics (MD) code LAMMPS is used to perform the calculations. OVITO is used to visualize the atomic structure before and after the creation of the interstitial. LAMMPS[1].
Author(s): Shakila Taylor*, Firas Akasheh*, Mark A. Tschopp, mark.tschopp@gatech.edu
Advisor(s): Firas Akasheh*, Mark A. Tschopp, mark.tschopp@gatech.edu
(*) Mechanical Engineering Department, Tuskegee University, Tuskegee, AL 36088
Introduction
Typically, metals are crystalline solids that have a periodic crystal structure such as FCC (face centered cubic) and BCC (body centered cubic), both of which are based on the cubic unit cell. The positions of atoms occur in repeating fixed positions, determined by the unit cell parameters. However, the arrangement of atoms in most crystalline materials is not perfect. The regular patterns are interrupted by crystallographic defects. Three types of defects that occur in metals are point, line, and planar defects. In this tutorial we focus on point defects, specifically on interstitials. Self-interstitial atoms are atoms of the same matrix material occupying positions which do not coincide with lattice points. Interstitials are important because they can modify the physical and chemical properties of materials.(https://en.wikipedia.org/wiki/Interstitial_defect).
Description of Simulation
First, LAMMPS generates a FCC copper (Cu) cell with a 8 x 8 x 8 dimension and a xyz orientation [100], [010], and [001] respectively with a total of 2048 atoms created. The boundaries of the cell structure are periodic in every direction. The input script file is written to simulate the insertion of one atom and calculate the resulting interstitial formation energy, Eif. First the number of atoms N in the cell is counted; afterwards the cell structure is relaxed and the initial energy E0 is computed; then an atom is inserted, the structure is relaxed again and the final energy Ef is computed. When all necessary values are obtained they are used to calculate the interstitial formation energy Eif. The equation used to calculate this energy is:
![E_i^f = E_f - [(N+1)/N] * E_0](img/interstitial_Equation.png)
Once the input file is written and run using LAMMPS, the file should display the initial and final energies, the number atoms and the interstitial formation energy.
LAMMPS input script for FCC
This input script was run using the December 21 2011 version of LAMMPS. The input script file was created and named Cu-interstitial.txt, but any name can be used. To run this script, store it in "Cu-interstitial.txt" and then use the "lmp_win_no-mpi.exe < Cu-interstitial.txt" in a Windows environment where "lmp_win_no-mpi.exe" refers to the LAMMPS executable.
# --------------- INITIALIZATION ------------------
clear
units metal
dimension 3
boundary p p p
atom_style atomic
# ------------------ ATOM DEFINITION -------------------
variable ao equal 3.615
lattice fcc 3.615
region simbox block -4 4 -4 4 -4 4
create_box 1 simbox
lattice fcc 3.615 orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
create_atoms 1 region simbox
# ------------------------ FORCE FIELDS -----------------------
pair_style eam/alloy
pair_coeff * * FeCuNi.eam.alloy Cu
#---------------------------Settings----------------------------
compute csym all centro/atom fcc
compute eng all pe/atom
compute eatoms all reduce sum c_eng
#----------------------Run Minimization-------------------------
reset_timestep 0
thermo 10
thermo_style custom step pe lx ly lz press pxx pyy pzz c_eatoms
dump 1 all custom 400 dump.relax.1.* id type xs ys zs c_csym c_eng
min_style cg
minimize 1e-15 1e-15 5000 5000
run 0
undump 1
#variable N equal count(all), counts the total number of atoms in the cell
#the total number of atoms is stored to the variable No
variable N equal count(all)
variable No equal $N
#variable E equal "c_eatoms" computes the initial energy of the cell system before the interstitial
#E is needed to store the initial energy of the system to the variable Eo
variable E equal "c_eatoms"
variable Eo equal $E
#---------------------------------------------------------------
variable r2 equal sqrt(${ao}^2+${ao}^2)/4
#r2 is the radius of the copper atom
#region select sphere 0 0 0 ${r2} units box
create_atoms 1 single 0 -1.8075 0 units box
# (0, -1.8075, 0) is the location of the inserted atom
# -1.8075 is half of the lattice (see figure 2)
#---------------------------------------------------------------
reset_timestep 0
thermo 10
thermo_style custom step pe lx ly lz press pxx pyy pzz c_eatoms
dump 1 all custom 400 dump.relax.2.* id type xs ys zs c_csym c_eng
min_style cg
minimize 1e-15 1e-15 5000 5000
#variable Ef equal "c_eatoms" computes the final energy of the cell system after the interstitial
#The final energy is stored to the variable Ef
variable Ef equal "c_eatoms"
variable Ei equal (${Ef}-((${No}+1)/${No})*${Eo})
#---------------------------------------------
######################################
# SIMULATION DONE
print "All done"
print "Total number of atoms = ${No}"
print "Initial energy of atoms = ${Eo}"
print "Final energy of atoms = ${Ef}"
print "Interstitial formation energy = ${Ei}"
Output
LAMMPS logfile
The log.lammps file should look like the one shown below.
LAMMPS (21 Dec 2011)
# --------------- INITIALIZATION ------------------
clear
units metal
dimension 3
boundary p p p
atom_style atomic
# ------------------ ATOM DEFINITION -------------------
variable ao equal 3.615
lattice fcc 3.615
Lattice spacing in x,y,z = 3.615 3.615 3.615
region simbox block -4 4 -4 4 -4 4
create_box 1 simbox
Created orthogonal box = (-14.46 -14.46 -14.46) to (14.46 14.46 14.46)
1 by 1 by 1 MPI processor grid
lattice fcc 3.615 orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
Lattice spacing in x,y,z = 3.615 3.615 3.615
create_atoms 1 region simbox
Created 2048 atoms
# ------------------------ FORCE FIELDS -----------------------
pair_style eam/alloy
pair_coeff * * FeCuNi.eam.alloy Cu
#---------------------------Settings----------------------------
compute csym all centro/atom fcc
compute eng all pe/atom
compute eatoms all reduce sum c_eng
#----------------------Run Minimization-------------------------
reset_timestep 0
thermo 10
thermo_style custom step pe lx ly lz press pxx pyy pzz c_eatoms
dump 1 all custom 400 dump.relax.1.* id type xs ys zs c_csym c_eng
min_style cg
minimize 1e-15 1e-15 5000 5000
WARNING: Resetting reneighboring criteria during minimization (min.cpp:167)
Memory usage per processor = 4.80068 Mbytes
Step PotEng Lx Ly Lz Press Pxx Pyy Pzz eatoms
0 -7250.3671 28.92 28.92 28.92 -86.036792 -86.036792 -86.036792 -86.036792 -7250.3671
1 -7250.3671 28.92 28.92 28.92 -86.036792 -86.036792 -86.036792 -86.036792 -7250.3671
Loop time of 0.062401 on 1 procs for 1 steps with 2048 atoms
Minimization stats:
Stopping criterion = linesearch alpha is zero
Energy initial, next-to-last, final =
-7250.36709989 -7250.36709989 -7250.36709989
Force two-norm initial, final = 2.85021e-013 2.85021e-013
Force max component initial, final = 7.10543e-015 7.10543e-015
Final line search alpha, max atom move = 0.5 3.55271e-015
Iterations, force evaluations = 1 2
Pair time (%) = 0.0156001 (24.9998)
Neigh time (%) = 0 (0)
Comm time (%) = 0.0156003 (25)
Outpt time (%) = 0 (0)
Other time (%) = 0.0312006 (50.0002)
Nlocal: 2048 ave 2048 max 2048 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Nghost: 5765 ave 5765 max 5765 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Neighs: 143360 ave 143360 max 143360 min
Histogram: 1 0 0 0 0 0 0 0 0 0
FullNghs: 286720 ave 286720 max 286720 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Total # of neighbors = 286720
Ave neighs/atom = 140
Neighbor list builds = 0
Dangerous builds = 0
run 0
WARNING: No fixes defined, atoms won't move (verlet.cpp:52)
Memory usage per processor = 4.11404 Mbytes
Step PotEng Lx Ly Lz Press Pxx Pyy Pzz eatoms
1 -7250.3671 28.92 28.92 28.92 -86.036792 -86.036792 -86.036792 -86.036792 -7250.3671
Loop time of 6.56582e-008 on 1 procs for 0 steps with 2048 atoms
Pair time (%) = 0 (0)
Neigh time (%) = 0 (0)
Comm time (%) = 0 (0)
Outpt time (%) = 0 (0)
Other time (%) = 6.56582e-008 (100)
Nlocal: 2048 ave 2048 max 2048 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Nghost: 5765 ave 5765 max 5765 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Neighs: 143360 ave 143360 max 143360 min
Histogram: 1 0 0 0 0 0 0 0 0 0
FullNghs: 286720 ave 286720 max 286720 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Total # of neighbors = 286720
Ave neighs/atom = 140
Neighbor list builds = 0
Dangerous builds = 0
undump 1
#variable N equal count(all), counts the total number of atoms in the cell
#the total number of atoms is stored to the variable No
variable N equal count(all)
variable No equal $N
variable No equal 2048
#variable E equal "c_eatoms" computes the initial energy of the cell system before the interstitial
#E is needed to store the initial energy of the system to the variable Eo
variable E equal "c_eatoms"
variable Eo equal $E
variable Eo equal -7250.3671
#---------------------------------------------------------------
variable r2 equal sqrt(${ao}^2+${ao}^2)/4
variable r2 equal sqrt(3.615^2+${ao}^2)/4
variable r2 equal sqrt(3.615^2+3.615^2)/4
#r2 is the radius of the copper atom
#region select sphere 0 0 0 ${r2} units box
create_atoms 1 single 0 -1.8075 0 units box
Created 1 atoms
# (0, -1.8075, 0) is the location of the inserted atom
# -1.8075 is half of the lattice (see figure 2)
#---------------------------------------------------------------------
reset_timestep 0
thermo 10
thermo_style custom step pe lx ly lz press pxx pyy pzz c_eatoms
dump 1 all custom 400 dump.relax.2.* id type xs ys zs c_csym c_eng
min_style cg
minimize 1e-15 1e-15 5000 5000
WARNING: Resetting reneighboring criteria during minimization (min.cpp:167)
Memory usage per processor = 4.80068 Mbytes
Step PotEng Lx Ly Lz Press Pxx Pyy Pzz eatoms
0 -7238.8086 28.92 28.92 28.92 2990.7332 2990.7332 2990.7332 2990.7332 -7238.8086
10 -7250.6389 28.92 28.92 28.92 1196.793 1196.793 1196.793 1196.793 -7250.6389
20 -7250.6531 28.92 28.92 28.92 1197.6886 1197.6886 1197.6886 1197.6886 -7250.6531
30 -7250.6532 28.92 28.92 28.92 1197.1798 1197.1798 1197.1798 1197.1798 -7250.6532
35 -7250.6532 28.92 28.92 28.92 1197.1709 1197.1709 1197.1709 1197.1709 -7250.6532
Loop time of 1.2162 on 1 procs for 35 steps with 2049 atoms
Minimization stats:
Stopping criterion = linesearch alpha is zero
Energy initial, next-to-last, final =
-7238.80861596 -7250.65321113 -7250.65321113
Force two-norm initial, final = 31.4309 0.000378851
Force max component initial, final = 12.8314 0.000156888
Final line search alpha, max atom move = 0.0625 9.80553e-006
Iterations, force evaluations = 35 125
Pair time (%) = 1.1694 (96.1519)
Neigh time (%) = 0 (0)
Comm time (%) = 0.0155988 (1.28258)
Outpt time (%) = -5.56465e-008 (-4.57543e-006)
Other time (%) = 0.0312018 (2.56551)
Nlocal: 2049 ave 2049 max 2049 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Nghost: 5765 ave 5765 max 5765 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Neighs: 143524 ave 143524 max 143524 min
Histogram: 1 0 0 0 0 0 0 0 0 0
FullNghs: 287468 ave 287468 max 287468 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Total # of neighbors = 287468
Ave neighs/atom = 140.297
Neighbor list builds = 0
Dangerous builds = 0
#variable Ef equal "c_eatoms" computes the final energy of the cell system after the interstitial
#The final energy is stored to the variable Ef
variable Ef equal "c_eatoms"
variable Ei equal (${Ef}-((${No}+1)/${No})*${Eo})
variable Ei equal (-7250.653211-((${No}+1)/${No})*${Eo})
variable Ei equal (-7250.653211-((2048+1)/${No})*${Eo})
variable Ei equal (-7250.653211-((2048+1)/2048)*${Eo})
variable Ei equal (-7250.653211-((2048+1)/2048)*-7250.3671)
#---------------------------------------------
######################################
# SIMULATION DONE
print "All done"
All done
print "Total number of atoms = ${No}"
Total number of atoms = 2048
print "Initial energy of atoms = ${Eo}"
Initial energy of atoms = -7250.3671
print "Final energy of atoms = ${Ef}"
Final energy of atoms = -7250.653211
print "Interstitial formation energy = ${Ei}"Interstitial formation energy = 3.254107311
Post-Processing
Visualization
The simulation can be visualized using OVITO (www.ovito.org; developer: Alexander Stukowski). To do so, first open the Ovito program and select the import data tab. Then go to the directory and file in which you ran the simulation. Locate the first dumpfile, which would be dump.relax.1.0 in this case. You can then double click on the file, or select the file, then click open. The image of the cell should appear on the screen, displayed in four views, Figure 1. This image will be that of the relaxed cell before introducing the vacancy. To view the cell after interstitial has been created, follow the same steps above to select the dump file at the end of the relaxation which followed the atom insertion (these files are labeled dump.relax.2.*. Once you open the new file, select one of the four views. Then open the add modification tab and from the drop down menu select Slice. Set the Normal (X) to 1 and set Normal (Y) and Normal (Z) to 0. Return to the add modification tab and select Color coding. When here select c_eng for the Property. Then select Rainbow for the Color gradient and click Adjust range. The interstitial should be visible in the center of the cell, colored by the energy surrounding the vacancy, see Figure 3.

Figure 1: Perspective view of the simulation cell with 2048 total atoms before a vacancy is created.
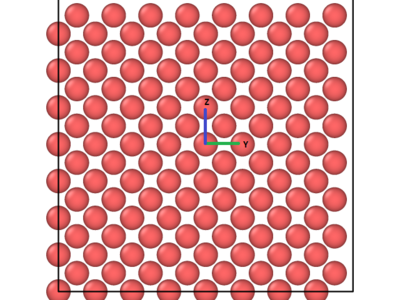
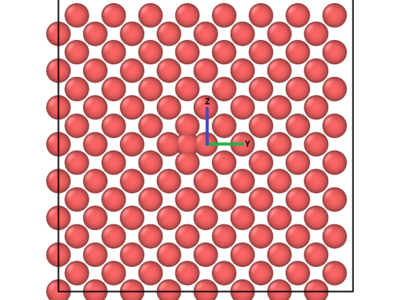
Figure 2: Before and after views of the ZY – plane where the interstitial is located
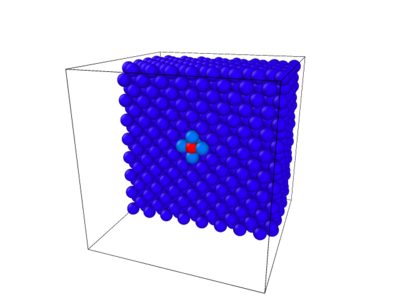
Figure 3: Slice through the center of the simulation cell showing the interstitial. Atoms are colored by the energy per atom. The red atom was inserted into the cell displaying the highest energy. The 4 light blue atoms surrounding the red, have a lower energy then the red because they were shifted outward as a result of the additional atom being forced into the cell.
Acknowledgments
We would like to acknowledge the support to this work by the National Science Foundation, HBCUUP-RIA program, Program Manager Dr. Claudia Rankins, Award No. HRD-1137587. Additionally, the technical and logistical support of CAVS and HPC2 of Mississippi State University is gratefully acknowledged.
References
- ↑ S. Plimpton, "Fast Parallel Algorithms for Short-Range Molecular Dynamics," J. Comp. Phys., 117, 1-19 (1995). (http://www.sciencedirect.com/science/article/pii/S002199918571039X)