
Integrated Computational Materials Engineering (ICME)
Grain boundary generation
Abstract
The objective of this research is to generate grain boundary structures over a wide range of degrees of freedom for future use in assessing how grain boundary degrees of freedom impact the properties of polycrystalline materials. For each grain boundary, this work uses a parallel molecular dynamics code, LAMMPS, with in-plane translations and atom deletion criteria to sample a large number of potential structures to find the global minimum energy grain boundary structure[1][2]. The results of this work are grain boundary structures that agree with previously calculated structures as well as experimentally-imaged HRTEM structures. Additionally, the grain boundary energies agree with previous calculated and measured energies. The significance of this research is that grain boundary properties play an important role in the properties of polycrystalline materials and this research enables future atomistic research investigating these properties.
Author(s): Mark A. Tschopp, Mark F. Horstemeyer
Corresponding Author: Mark Tschopp

Movie showing the evolution of grain boundary structure for aluminum <100> symmetric tilt grain boundaries as a function of misorientation angle.
Methodology
For each grain boundary structure, the crystal orientations and their relationship to the grain boundary plane -- five degrees of freedom -- are often used to describe the grain boundary character. A three-dimensional periodic simulation cell is used for these calculations (see Fig. 1), whereby there are two crystal orientations and two grain boundaries (one in the middle and one at the top/bottom). This simulation cell allows the user to manipulate the five grain boundary degrees of freedom to investigate their relationship on properties. The distance between the two boundaries must be large enough for the grain boundary energy to converge, i.e., in the input script below, a distance of 12 nm was used. Figure 2. This is a plot of the accessibility of each grain boundary structure for an asymmetric tilt grain boundary. For complex boundaries, a large number of boundaries may need to be sampled to find the "global" minimum energy boundary.
In many cases, at the atomistic level, the minimum energy structure cannot be found by merely adjoining the two crystal lattices in the simulation cell. The relative translation of the two crystal lattices with respect to each other is important as well as the number of atoms within the grain boundary region. Therefore, to find the minimum energy structure, a large number of configurations with different relative translations is vital to finding the stable minimum energy structure and not just a metastable grain boundary structure. Figure 2 shows an example for an asymmetric tilt grain boundary, where the minimum energy structure was only found (accessed) 8.76% of the time with various translations. In more complex boundaries, the accessibility may be less than 0.1%.
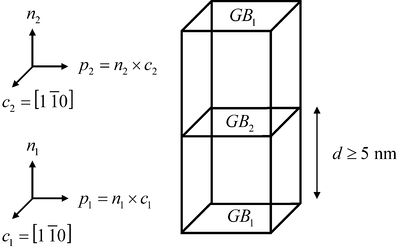
Figure 1. An example of the bicrystal simulation cell used for the grain boundary structure calculations. In this particular example, the grain boundary is a <110> tilt grain boundary. (click on the image to enlarge).
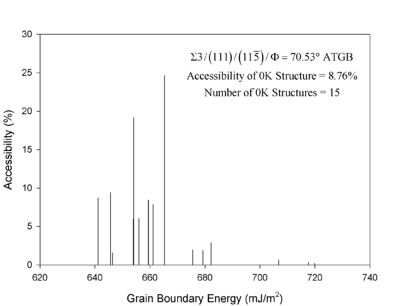
Figure 2. This is a plot of the accessibility of each grain boundary structure for an asymmetric tilt grain boundary. For complex boundaries, a large number of boundaries may need to be sampled to find the "global" minimum energy boundary. (click on the image to enlarge).
The following input script shows how multiple translations and an atom deletion criteria are used to calculate the minimum energy structure. This input script for LAMMPS[3] can be called with a command of the form, "lmp_exe < input.script." This script contains loops over x-translations, z-translations, and atom overlap distances (an atom is deleted when an atom pair with a nearest neighbor distance is less than this distance). The unique minimum energy structures are saved as a dump file with the energy appended to the filename in a new folder specified by the 'gbname' variable. The dump files can then be easily scanned through for the global minimum energy structure.
Material Model
Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS)
Input Data
See LAMMPS Input Deck for Grain boundary generation
Results
This methodology has resulted in structures that agree with HRTEM images and grain boundary energies that agree with experimentally-measured grain boundary energies. Figure 3 shows an example plot of the grain boundary energy versus inclination angle for a complex grain boundaries in Cu (Sigma 3 asymmetric grain boundaries). This system of boundaries displays a phase transformation at the boundary to the orthorhombic 9R phase for inclination angles of 70-90 degrees. The methodology used above agrees nicely with experimental results and previous atomistic calculations of Wolf and coworkers.
Al <100> symmetric tilt grain boundaries.

Al <110> symmetric tilt grain boundaries.
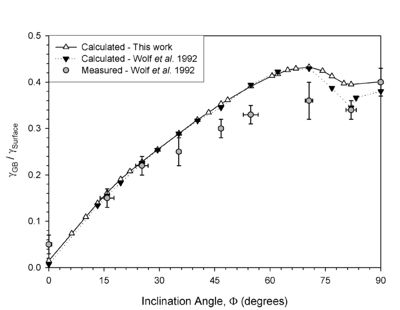
Figure 3. Plot of the grain boundary energy as a function of inclination angle for asymmetric tilt grain boundaries in Cu.

Cu <100> symmetric tilt grain boundaries.

Cu <110> symmetric tilt grain boundaries.
Acknowledgments
M.A. Tschopp would like to acknowledge funding provided under an NSF graduate fellowship for the initial work. Continued funding for investigating structure-property relationships in grain boundaries under the NEAMS (Nuclear Energy Advanced Modeling and Simulation) program is also acknowledged.
References
The initial methodology was used in the following papers:
- ↑ Tschopp, M. A., & McDowell, D.L. (2007). Structures and energies of Sigma3 asymmetric tilt grain boundaries in Cu and Al. Philosophical Magazine, 87, 3147-3173 (https://dx.doi.org/10.1080/14786430701455321).
- ↑ Tschopp, M. A., & McDowell, D.L. (2007). Asymmetric tilt grain boundary structure and energy in copper and aluminum. Philosophical Magazine, 87, 3871-3892 (https://dx.doi.org/10.1016/j.commatsci.2010.02.003).
- ↑ S. Plimpton, "Fast Parallel Algorithms for Short-Range Molecular Dynamics," J. Comp. Phys., 117, 1-19 (1995).