
Integrated Computational Materials Engineering (ICME)
LAMMPS Help3
Abstract
The objective of this tutorial is to generate a symmetric tilt grain boundary in LAMMPS. This tutorial can serve as a precursor to more advanced techniques, whereby in-plane translations and atom deletion criteria are used to sample a large number of potential structures to find the global minimum energy grain boundary structure[1][2].
Author(s): Mark A. Tschopp, Nayeon Lee
Corresponding Author: mark.tschopp@gatech.edu
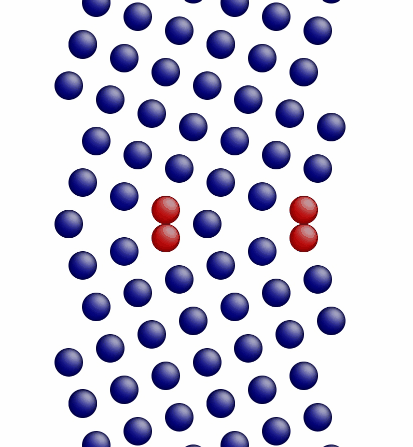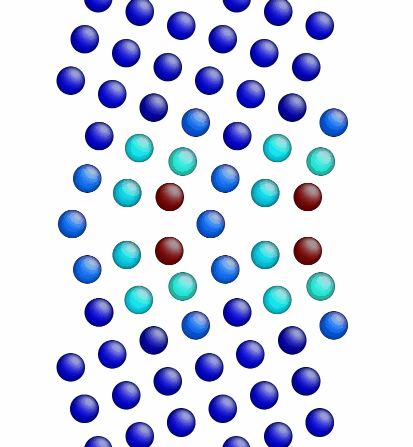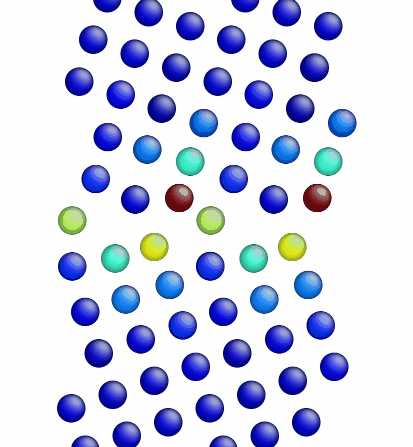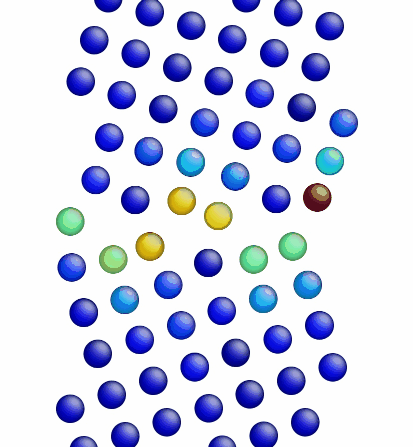
Movie showing the minimization of the grain boundary structure for an aluminum Sigma5 (310) symmetric tilt grain boundary.
Methodology
The following input script shows how multiple translations and an atom deletion criteria are used to calculate the minimum energy structure. This input script for LAMMPS[3] can be called with a command of the form, "lmp_exe < input.script." This script contains loops over x-translations, z-translations, and atom overlap distances (an atom is deleted when an atom pair with a nearest neighbor distance is less than this distance). The unique minimum energy structures are saved as a dump file with the energy appended to the filename in a new folder specified by the 'gbname' variable. The dump files can then be easily scanned through for the global minimum energy structure. Simulations performed on Aug 2014 LAMMPS version.
LAMMPS Input file
# LAMMPS Input File for Grain Boundaries
# Mark Tschopp, Dec2009
# This file will generate a single Sigma5(310) STGB
# ---------- Initialize Simulation ---------------------
clear
units metal
dimension 3
boundary p p p
atom_style atomic
# ---------- Create Atomistic Structure ---------------------
lattice fcc 4.05
region whole block 0.000000 12.807225 -64.0361225 64.0361225 0.000000 4.050000 units box
create_box 2 whole
region upper block INF INF 0.000000 64.0361225 INF INF units box
lattice fcc 4.05 orient x 0 3 1 orient y 0 -1 3 orient z 1 0 0
create_atoms 1 region upper
region lower block INF INF -64.0361225 0.000000 INF INF units box
lattice fcc 4.05 orient x 0 3 -1 orient y 0 1 3 orient z 1 0 0
create_atoms 2 region lower
group upper type 1
group lower type 2
# ---------- Define Interatomic Potential ---------------------
pair_style eam/alloy
pair_coeff * * Al99.eam.alloy Al Al
neighbor 2.0 bin
neigh_modify delay 10 check yes
# ---------- Displace atoms and delete overlapping atoms ---------------------
displace_atoms upper move 0 0 0 units lattice
delete_atoms overlap 0.35 lower upper
# ---------- Define Settings ---------------------
compute csym all centro/atom fcc
compute eng all pe/atom
compute eatoms all reduce sum c_eng
# ---------- Run Minimization ---------------------
reset_timestep 0
thermo 10
thermo_style custom step pe lx ly lz press pxx pyy pzz c_eatoms
dump 1 all cfg 25 dump.sig5_minimization_*.cfg mass type xs ys zs c_csym c_eng fx fy fz
dump_modify 1 element Al Al
min_style cg
minimize 1e-15 1e-15 5000 5000
undump 1
# ---------- Run Minimization 2---------------------
# Now allow the box to expand/contract perpendicular to the grain boundary
reset_timestep 0
thermo 10
thermo_style custom step pe lx ly lz press pxx pyy pzz c_eatoms
fix 1 all box/relax y 0 vmax 0.001
min_style cg
minimize 1e-15 1e-15 5000 5000
# ---------- Calculate GB Energy ---------------------
variable minimumenergy equal -3.360000
variable esum equal "v_minimumenergy * count(all)"
variable xseng equal "c_eatoms - (v_minimumenergy * count(all))"
variable gbarea equal "lx * lz * 2"
variable gbe equal "(c_eatoms - (v_minimumenergy * count(all)))/v_gbarea"
variable gbemJm2 equal ${gbe}*16021.7733
variable gbernd equal round(${gbemJm2})
print "GB energy is ${gbemJm2} mJ/m^2"
# ---------- Dump data into Data file -------------
reset_timestep 0
dump 1 all cfg 10000 dump.al_sig5_310_*.cfg mass type xs ys zs c_csym c_eng fx fy fz
dump_modify 1 element Al Al
minimize 1e-15 1e-15 5000 5000
undump 1
write_restart restart.al_sig5_310_stgb
print "All done"
LAMMPS Logfile
Here is an example logfile that is obtained after running this script.
LAMMPS (13 Jul 2010)
# LAMMPS Input File for Grain Boundaries
# Mark Tschopp, Dec2009
# This file will generate a single Sigma5(310) STGB
# ---------- Initialize Simulation ---------------------
clear
units metal
dimension 3
boundary p p p
atom_style atomic
# ---------- Create Atomistic Structure ---------------------
lattice fcc 4.05
Lattice spacing in x,y,z = 4.05 4.05 4.05
region whole block 0.000000 12.807225 -64.0361225 64.0361225 0.000000 4.050000 units box
create_box 2 whole
Created orthogonal box = (0 -64.0361 0) to (12.8072 64.0361 4.05)
1 by 1 by 1 processor grid
region upper block INF INF 0.000000 64.0361225 INF INF units box
lattice fcc 4.05 orient x 0 3 1 orient y 0 -1 3 orient z 1 0 0
Lattice spacing in x,y,z = 5.12289 5.12289 4.05
create_atoms 1 region upper
Created 200 atoms
region lower block INF INF -64.0361225 0.000000 INF INF units box
lattice fcc 4.05 orient x 0 3 -1 orient y 0 1 3 orient z 1 0 0
Lattice spacing in x,y,z = 5.12289 5.12289 4.05
create_atoms 2 region lower
Created 200 atoms
group upper type 1
200 atoms in group upper
group lower type 2
200 atoms in group lower
# ---------- Define Interatomic Potential ---------------------
pair_style eam/alloy
pair_coeff * * Al99.eam.alloy Al Al
neighbor 2.0 bin
neigh_modify delay 10 check yes
# ---------- Displace atoms and delete overlapping atoms ---------------------
displace_atoms upper move 0 0 0 units lattice
delete_atoms overlap 0.35 lower upper
Deleted 2 atoms, new total = 398
# ---------- Define Settings ---------------------
compute csym all centro/atom fcc
compute eng all pe/atom
compute eatoms all reduce sum c_eng
# ---------- Run Minimization ---------------------
reset_timestep 0
thermo 10
thermo_style custom step pe lx ly lz press pxx pyy pzz c_eatoms
dump 1 all cfg 25 dump.sig5_minimization_*.cfg id type xs ys zs c_csym c_eng fx fy fz
dump_modify 1 element Al Al
min_style cg
minimize 1e-15 1e-15 5000 5000
Memory usage per processor = 3.58369 Mbytes
Step PotEng Lx Ly Lz Press Pxx Pyy Pzz eatoms
0 -1318.9843 12.807225 128.07225 4.05 875.32429 -2675.7752 7333.2608 -2031.5128 -1318.9843
10 -1329.7948 12.807225 128.07225 4.05 4786.2646 2230.1043 9306.1111 2822.5783 -1329.7948
20 -1330.138 12.807225 128.07225 4.05 5741.5477 3251.6199 10395.111 3577.9118 -1330.138
30 -1330.2064 12.807225 128.07225 4.05 5762.2257 3246.6439 10519.567 3520.4656 -1330.2064
40 -1330.2128 12.807225 128.07225 4.05 5812.6504 3332.1043 10542.913 3562.9335 -1330.2128
50 -1330.2136 12.807225 128.07225 4.05 5811.3217 3335.8029 10533.825 3564.3374 -1330.2136
766 -1333.6024 12.807225 128.07225 4.05 -6.3617301 -262.3388 1197.774 -954.5204 -1333.6024
Loop time of 5.19081 on 1 procs for 766 steps with 398 atoms
Minimization stats:
Stopping criterion = linesearch alpha is zero
Energy initial, next-to-last, final =
-1318.98429985 -1333.60243356 -1333.60243356
Force two-norm initial, final = 17.5886 0.000221422
Force max component initial, final = 6.25036 3.74708e-05
Final line search alpha, max atom move = 0.25 9.36769e-06
Iterations, force evaluations = 766 2310
Pair time (%) = 4.35802 (83.9565)
Neigh time (%) = 0.00490403 (0.0944753)
Comm time (%) = 0.297343 (5.72827)
Outpt time (%) = 0.391773 (7.54745)
Other time (%) = 0.138767 (2.67332)
Nlocal: 398 ave 398 max 398 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Nghost: 5281 ave 5281 max 5281 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Neighs: 27704 ave 27704 max 27704 min
Histogram: 1 0 0 0 0 0 0 0 0 0
FullNghs: 55442 ave 55442 max 55442 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Total # of neighbors = 55442
Ave neighs/atom = 139.302
Neighbor list builds = 3
Dangerous builds = 0
undump 1
# ---------- Run Minimization 2---------------------
# Now allow the box to expand/contract perpendicular to the grain boundary
reset_timestep 0
thermo 10
thermo_style custom step pe lx ly lz press pxx pyy pzz c_eatoms
fix 1 all box/relax y 0 vmax 0.001
min_style cg
minimize 1e-15 1e-15 5000 5000
Memory usage per processor = 3.43888 Mbytes
Step PotEng Lx Ly Lz Press Pxx Pyy Pzz eatoms
0 -1333.6024 12.807225 128.07225 4.05 -6.3617301 -262.3388 1197.774 -954.5204 -1333.6024
10 -1333.605 12.807225 128.20832 4.05 -841.94577 -891.13967 -20.783769 -1613.9139 -1333.605
20 -1333.605 12.807225 128.20608 4.05 -826.51441 -879.30405 3.3043329 -1603.5435 -1333.605
30 -1333.605 12.807225 128.20654 4.05 -828.43998 -880.95627 2.999739 -1607.3634 -1333.605
40 -1333.605 12.807225 128.20729 4.05 -832.39474 -884.11935 -0.96690522 -1612.098 -1333.605
42 -1333.605 12.807225 128.20679 4.05 -829.31937 -881.81499 3.5751362 -1609.7182 -1333.605
Loop time of 0.797956 on 1 procs for 42 steps with 398 atoms
Minimization stats:
Stopping criterion = linesearch alpha is zero
Energy initial, next-to-last, final =
-1333.60243356 -1333.60504971 -1333.60504971
Force two-norm initial, final = 4.96626 0.0149609
Force max component initial, final = 4.96626 0.0148234
Final line search alpha, max atom move = 6.91964e-05 1.02572e-06
Iterations, force evaluations = 42 331
Pair time (%) = 0.674209 (84.492)
Neigh time (%) = 0 (0)
Comm time (%) = 0.0393722 (4.93413)
Outpt time (%) = 0.000830889 (0.104127)
Other time (%) = 0.0835438 (10.4697)
Nlocal: 398 ave 398 max 398 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Nghost: 5233 ave 5233 max 5233 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Neighs: 27721 ave 27721 max 27721 min
Histogram: 1 0 0 0 0 0 0 0 0 0
FullNghs: 55442 ave 55442 max 55442 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Total # of neighbors = 55442
Ave neighs/atom = 139.302
Neighbor list builds = 0
Dangerous builds = 0
# ---------- Calculate GB Energy ---------------------
variable minimumenergy equal -3.360000
variable esum equal "v_minimumenergy * count(all)"
variable xseng equal "c_eatoms - (v_minimumenergy * count(all))"
variable gbarea equal "lx * lz * 2"
variable gbe equal "(c_eatoms - (v_minimumenergy * count(all)))/v_gbarea"
variable gbemJm2 equal ${gbe}*16021.7733
variable gbemJm2 equal 0.03542512656*16021.7733
variable gbernd equal round(${gbemJm2})
variable gbernd equal round(567.5733469)
print "GB energy is ${gbemJm2} mJ/m^2"
GB energy is 567.5733469 mJ/m^2
# ---------- Dump data into Data file -------------
reset_timestep 0
dump 1 all cfg 10000 dump.al_sig5_310_*.cfg id type xs ys zs c_csym c_eng fx fy fz
dump_modify 1 element Al Al
minimize 1e-15 1e-15 5000 5000
Memory usage per processor = 3.58369 Mbytes
Step PotEng Lx Ly Lz Press Pxx Pyy Pzz eatoms
0 -1333.605 12.807225 128.20679 4.05 -829.31937 -881.81499 3.5751362 -1609.7182 -1333.605
2 -1333.605 12.807225 128.20704 4.05 -830.86046 -882.97143 1.3125102 -1610.9225 -1333.605
Loop time of 0.0632482 on 1 procs for 2 steps with 398 atoms
Minimization stats:
Stopping criterion = linesearch alpha is zero
Energy initial, next-to-last, final =
-1333.60504971 -1333.60504973 -1333.60504973
Force two-norm initial, final = 0.0149764 0.00581102
Force max component initial, final = 0.014839 0.0054477
Final line search alpha, max atom move = 0.000358523 1.95313e-06
Iterations, force evaluations = 2 20
Pair time (%) = 0.0428157 (67.6948)
Neigh time (%) = 0 (0)
Comm time (%) = 0.00247025 (3.90566)
Outpt time (%) = 0 (0)
Other time (%) = 0.0179622 (28.3996)
Nlocal: 398 ave 398 max 398 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Nghost: 5233 ave 5233 max 5233 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Neighs: 27721 ave 27721 max 27721 min
Histogram: 1 0 0 0 0 0 0 0 0 0
FullNghs: 55442 ave 55442 max 55442 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Total # of neighbors = 55442
Ave neighs/atom = 139.302
Neighbor list builds = 0
Dangerous builds = 0
undump 1
write_restart restart.al_sig5_310_stgb
print "All done"
All done
The grain boundary energy is calculated as 567.57 mJ/m2. The final configuration is stored in a restart file for future use in predicting properties of grain boundaries. Additionally, files with atomic coordinates, energies, and centrosymmetry values are dumped for post-processing.
Images and movies
Figure 1 shows a movie of minimization of the grain boundary structure for the input script show above. Figure 2 shows a movie of minimization of the grain boundary structure for a modification of the input script above. Here, the "delete_atoms overlap 0.35 lower upper" command has been modified to "delete_atoms overlap 1.5 lower upper". This eliminates two atoms that are too close to each other near the grain boundary interface.
Figure 1. Movie showing the minimization of the grain boundary structure for an aluminum Sigma5 (310) symmetric tilt grain boundary.
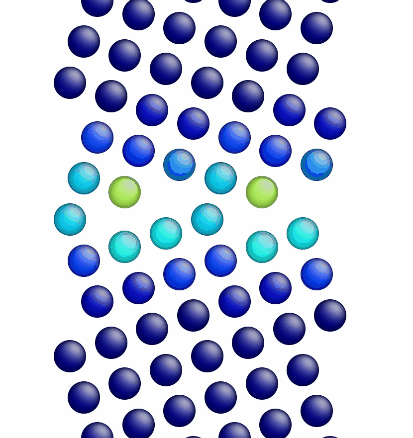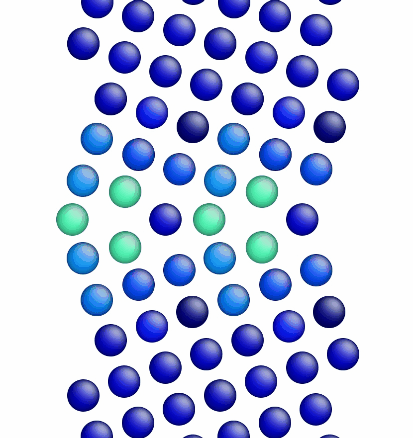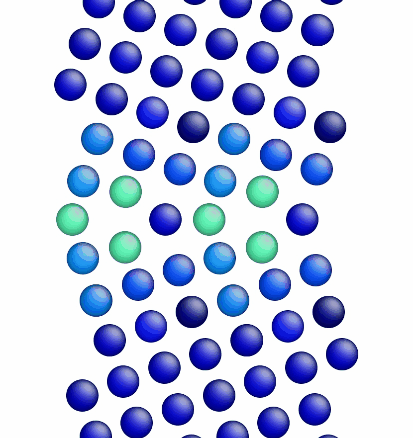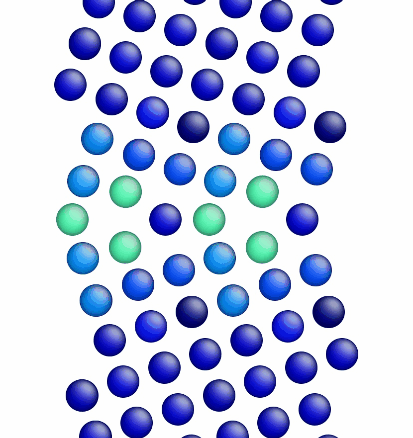
Figure 2. Movie showing the minimization of the grain boundary structure for an aluminum Sigma5 (310) symmetric tilt grain boundary.
Figure 3 shows the image corresponding to the input script shown above. Notice how there are two atoms that are very close to each other at the boundary in Figure 3 (shown in red, colored by potential energy, i.e., high energy atoms). The final structures are shown in Figures 4 and 5 for the input script above and the modified input script, respectively. Interestingly, with this potential, the structure in Figure 4 is actually the lower energy grain boundary structure. The different structures for this one simple grain boundary may impart slightly different properties as well. Often, the convention is to use the minimum energy grain boundary structure for properties. However, some recent research has also explored metastable higher energy grain boundary structures and their impact on properties as well.
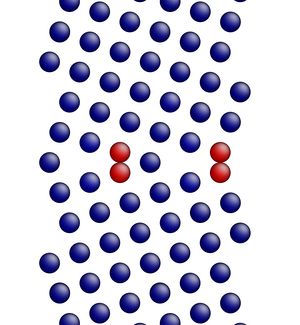
Figure 3. Image showing the grain boundary structure prior to minimization.
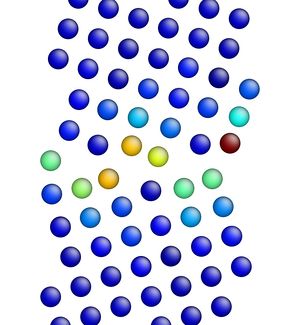
Figure 4. Image showing the grain boundary structure after minimization (overlap distance equals 0.35).
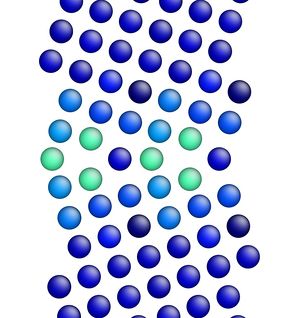
Figure 5. Image showing the grain boundary structure after minimization (overlap distance equals 1.50).
Acknowledgments
M.A. Tschopp would like to acknowledge funding provided under an NSF graduate fellowship for the initial work. Continued funding for investigating structure-property relationships in grain boundaries under the NEAMS (Nuclear Energy Advanced Modeling and Simulation) program is also acknowledged.
References
The initial methodology was used in the following papers:- ↑ Tschopp, M. A., & McDowell, D.L. (2007). Structures and energies of Sigma3 asymmetric tilt grain boundaries in Cu and Al. Philosophical Magazine, 87, 3147-3173 (http://dx.doi.org/10.1080/14786430701455321).
- ↑ Tschopp, M. A., & McDowell, D.L. (2007). Asymmetric tilt grain boundary structure and energy in copper and aluminum. Philosophical Magazine, 87, 3871-3892 (http://dx.doi.org/10.1016/j.commatsci.2010.02.003).
- ↑ S. Plimpton, "Fast Parallel Algorithms for Short-Range Molecular Dynamics," J. Comp. Phys., 117, 1-19 (1995).