
Integrated Computational Materials Engineering (ICME)
Uniaxial Compression
Abstract
This example shows how to run an atomistic simulation of uniaxial compressive loading of an aluminum single crystal oriented in the <100> direction. It also compares results of simulations with 4,000, 32,000, and 108,000 atoms. This example uses a parallel molecular dynamics code, LAMMPS[1]. These scripts were initially used to study dislocation nucleation in single crystal aluminum and copper[2][3][4].
Author(s): Mark A. Tschopp, Nathan R. Rhodes
Input
Description of Simulation
This molecular dynamics simulation first generates a simulation cell with fcc atoms with <100> orientations in the x, y, and z-directions. For this example, the simulation cell size is 10 lattice units in each direction for 4,000 total atoms, 20 units for 32,000 atoms, and 30 units for 108,000 atoms. Larger simulation cell sizes should be used to converge the dislocation nucleation stress values and to not influence the dislocation nucleation mechanism. The potential used in the Mishin et al. (1999) aluminum potential[5]. The equilibration step allows the lattice to expand to a temperature of 300 K with a pressure of 0 bar at each simulation cell boundary. Then, the simulation cell is deformed in the x-direction at a strain rate of 1010 s-1, while the lateral boundaries are controlled using the NPT equations of motion to zero pressure. The stress and strain values are output to a separate file, which can be imported in a graphing application for plotting. The cfg dump files include the x, y, and z coordinates, the centrosymmetry values, the potential energies, and forces for each atom. This can be directly visualized using AtomEye[6].
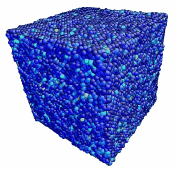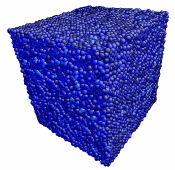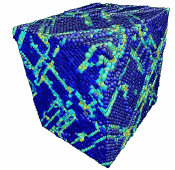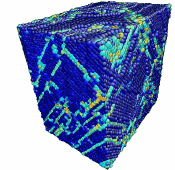
Movie showing compressive deformation of single crystal of 108,000 aluminum atoms loaded in the <100> direction at a strain rate of 1010 s-1 and a temperature of 300 K.
LAMMPS input script
This input script was run using the Aug 2015 version of LAMMPS. Changes in some commands in more recent versions may require revision of the input script. This script runs the simulation with 4,000 atoms. To change the number of atoms, alter the numbers (currently 10) after the "region" command under "Atom Definition." To run this script, store it in "in.comp.txt" and then use "lmp_exe < in.comp.txt" in a UNIX environment where "lmp_exe" refers to the LAMMPS executable.
Notice that the only difference, besides file names, between this script and the script for uniaxial tension is the negative in the definition of variable "srate1."
# Input file for uniaxial compressive loading of single crystal aluminum
# Mark Tschopp, November 2010
# ------------------------ INITIALIZATION ----------------------------
units metal
dimension 3
boundary p p p
atom_style atomic
variable latparam equal 4.05
# ----------------------- ATOM DEFINITION ----------------------------
lattice fcc ${latparam}
region whole block 0 10 0 10 0 10
create_box 1 whole
region upper block INF INF INF INF INF INF units box
lattice fcc ${latparam} orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
create_atoms 1 region upper
# ------------------------ FORCE FIELDS ------------------------------
pair_style eam/alloy
pair_coeff * * Al99.eam.alloy Al
# ------------------------- SETTINGS ---------------------------------
compute csym all centro/atom fcc
compute peratom all pe/atom
######################################
# EQUILIBRATION
reset_timestep 0
timestep 0.001
velocity all create 300 12345 mom yes rot no
fix 1 all npt temp 300 300 1 iso 0 0 1 drag 1
# Set thermo output
thermo 1000
thermo_style custom step lx ly lz press pxx pyy pzz pe temp
# Run for at least 10 picosecond (assuming 1 fs timestep)
run 20000
unfix 1
# Store final cell length for strain calculations
variable tmp equal "lx"
variable L0 equal ${tmp}
print "Initial Length, L0: ${L0}"
######################################
# DEFORMATION
reset_timestep 0
fix 1 all npt temp 300 300 1 y 0 0 1 z 0 0 1 drag 1
variable srate equal 1.0e10
variable srate1 equal "-v_srate / 1.0e12"
fix 2 all deform 1 x erate ${srate1} units box remap x
# Output strain and stress info to file
# for units metal, pressure is in [bars] = 100 [kPa] = 1/10000 [GPa]
# p2, p3, p4 are in GPa
variable strain equal "(lx - v_L0)/v_L0"
variable p1 equal "v_strain"
variable p2 equal "-pxx/10000"
variable p3 equal "-pyy/10000"
variable p4 equal "-pzz/10000"
fix def1 all print 100 "${p1} ${p2} ${p3} ${p4}" file Al_comp_100.def1.txt screen no
# Use cfg for AtomEye
dump 1 all cfg 250 dump.comp_*.cfg mass type xs ys zs c_csym c_peratom fx fy fz
dump_modify 1 element Al
# Display thermo
thermo 1000
thermo_style custom step v_strain temp v_p2 v_p3 v_p4 ke pe press
run 20000
######################################
# SIMULATION DONE
print "All done"
Output
LAMMPS logfile
The log.lammps file should look like this below. Notice that the 20 ps (20,000 fs) equilibration step brought the temperature of the simulation cell up to 300 K. The corresponding deformation went up to a strain of 0.2. The thermo command was set for every 1000 timesteps, so any plots should be made from the data included in the datafile.
LAMMPS (5 Nov 2010)
# Input file for uniaxial compressive loading of single crystal aluminum
#Mark Tschopp, November 2010
# ----- INITIALIZATION -----
units metal
dimension 3
boundary p p p
atom_style atomic
variable latparam equal 4.05
# ----- ATOM DEFINITION -----
lattice fcc ${latparam}
lattice fcc 4.05
Lattice spacing in x,y,z = 4.05 4.05 4.05
region whole block 0 10 0 10 0 10
create_box 1 whole
Created orthogonal box = (0 0 0) to (40.5 40.5 40.5)
2 by 2 by 2 processor grid
region upper block INF INF INF INF INF INF units box
lattice fcc ${latparam} orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
lattice fcc 4.05 orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
Lattice spacing in x,y,z = 4.05 4.05 4.05
create_atoms 1 region upper
Created 4000 atoms
# ----- FORCE FIELDS -----
pair_style eam/alloy
pair_coeff * * Al99.eam.alloy Al
# -----SETTINGS -----
compute csym all centro/atom fcc
compute peratom all pe/atom
# ----- EQUILIBRATION -----
reset_timestep 0
timestep 0.001
velocity all create 300 12345 mom yes rot no
fix 1 all npt temp 300 300 1 iso 0 0 1 drag 1
# Set Thermo Input
thermo 1000
thermo_style custom step lx ly lz press pxx pyy pzz pe temp
# Run Time (At least 10 picoseconds)
run 20000
Memory usage per processor = 2.55571 Mbytes
Step Lx Ly Lz Press Pxx Pyy Pzz PotEng Temp
0 40.5 40.5 40.5 2496.1233 2446.9902 2534.6541 2506.7256 -13440 300
1000 40.558342 40.558342 40.558342 731.50829 755.55351 696.37101 742.60035 -13363.301 169.49172
2000 40.574199 40.574199 40.574199 106.78935 -46.253007 296.26883 70.352219 -13356.097 178.17311
3000 40.579471 40.579471 40.579471 314.81803 314.71612 292.22553 337.51245 -13347.025 183.623
4000 40.586697 40.586697 40.586697 107.84524 52.829414 112.73441 157.9719 -13340.823 194.61149
5000 40.593224 40.593224 40.593224 172.67874 158.86784 199.13978 160.02861 -13334.364 204.85854
6000 40.601567 40.601567 40.601567 56.953653 -6.2830484 -16.301111 193.44512 -13327.55 213.93605
7000 40.60782 40.60782 40.60782 31.891969 35.437938 63.396951 -3.1589825 -13320.86 222.48071
8000 40.616908 40.616908 40.616908 -244.41321 -212.11716 -305.70692 -215.41555 -13317.384 236.254
9000 40.618876 40.618876 40.618876 13.561356 111.07353 -29.999797 -40.389663 -13313.071 247.18002
10000 40.625506 40.625506 40.625506 -125.06329 -105.4566 -93.514599 -176.21866 -13308.875 256.93613
11000 40.631525 40.631525 40.631525 -75.690987 141.36136 14.068057 -382.50238 -13303.582 263.00631
12000 40.631815 40.631815 40.631815 79.38005 63.796397 144.76416 29.579595 -13299.23 269.23326
13000 40.637408 40.637408 40.637408 -152.9113 -399.37081 -31.484802 -27.878283 -13298.162 280.0582
14000 40.636962 40.636962 40.636962 237.09239 58.799334 196.78768 455.69014 -13294.166 283.43969
15000 40.641455 40.641455 40.641455 105.44503 114.55343 142.4659 59.315739 -13291.527 287.73561
16000 40.643072 40.643072 40.643072 82.986611 -130.36308 288.9926 90.330318 -13288.384 289.39655
17000 40.64789 40.64789 40.64789 -12.310673 -112.67891 -132.8659 208.6128 -13289.081 296.89271
18000 40.644319 40.644319 40.644319 269.36896 180.82571 218.97472 408.30644 -13286.862 297.25615
19000 40.646772 40.646772 40.646772 166.57104 20.881491 -79.807885 558.63952 -13287.577 301.80321
20000 40.649126 40.649126 40.649126 116.57534 -108.86953 156.54682 302.04873 -13284.646 298.01867
Loop time of 79.3677 on 8 procs for 20000 steps with 4000 atoms
Pair time (%) = 63.798 (80.3828)
Neigh time (%) = 0 (0)
Comm time (%) = 12.2773 (15.4689)
Outpt time (%) = 0.00290701 (0.00366271)
Other time (%) = 3.28956 (4.1447)
Nlocal: 500 ave 500 max 500 min
Histogram: 8 0 0 0 0 0 0 0 0 0
Nghost: 2930 ave 2930 max 2930 min
Histogram: 8 0 0 0 0 0 0 0 0 0
Neighs: 35000 ave 36430 max 33540 min
Histogram: 2 0 1 1 0 0 1 1 0 2
FullNghs: 0 ave 0 max 0 min
Histogram: 8 0 0 0 0 0 0 0 0 0
Total # of neighbors = 280000
Ave neighs/atom = 70
Neighbor list builds = 0
Dangerous builds = 0
unfix 1
# Store final cell length for strain calculations
variable tmp equal "lx"
variable L0 equal ${tmp}
variable L0 equal 40.64912642
print "Initial Length, L0: ${L0}"
Initial Length, L0: 40.64912642
# -----DEFORMATION -----
reset_timestep 0
fix 1 all npt temp 300 300 1 y 0 0 1 z 0 0 1 drag 1
variable srate equal 1e10
#The only difference between this compressive example and
#the previous tensile example is the negative sign in the
#definition of srate1, seen below.
variable srate1 equal "-v_srate / 1e12"
fix 2 all deform 1 x erate ${srate1} units box remap x
fix 2 all deform 1 x erate -0.01 units box remap x
# Output strain and stress information to file.
# For metal units, pressure is [bars] = 100 [kPa] = 1/10000 [GPa]
# p2, p3, and p4 are in GPa
variable strain equal "(lx - v_L0) / v_L0"
variable p1 equal "v_strain"
variable p2 equal "-pxx/10000"
variable p3 equal "-pyy/10000"
variable p4 equal "-pzz/10000"
fix def1 all print 100 "${p1} ${p2} ${p3} ${p4}" file Al_comp.def1.txt screen no
# Dump to cfg for AtomEye post processing
dump 1 all cfg 250 dump.comp_*.cfg id type xs ys zs c_csym c_peratom fx fy fz
dump_modify 1 element Al
# Display thermo information
thermo 1000
thermo_style custom step v_strain temp v_p2 v_p3 v_p4 ke pe press
run 20000
Memory usage per processor = 2.8622 Mbytes
Step strain Temp p2 p3 p4 KinEng PotEng Press
0 -8.7965858e-11 298.01867 0.010886953 -0.015654682 -0.030204873 154.04923 -13284.646 116.57534
1000 -0.01 302.82586 -0.61033601 0.054190295 -0.0032380699 156.53411 -13286.645 1864.6126
2000 -0.02 298.61071 -1.2849368 -0.00313581 0.026906426 154.35526 -13281.302 4203.8871
3000 -0.03 302.18611 -1.8380212 -0.011375474 0.0087206074 156.20342 -13277.349 6135.5869
4000 -0.04 297.31028 -2.4628066 -0.0095117003 -0.026443669 153.68305 -13266.262 8329.2065
5000 -0.05 294.28788 -3.0927297 -0.0021071467 -0.050407399 152.12073 -13253.309 10484.147
6000 -0.06 301.6804 -3.7194292 -0.01909606 0.018996497 155.94201 -13242.696 12398.429
7000 -0.07 298.64768 -4.4073614 0.019050091 0.051288755 154.37437 -13223.403 14456.742
8000 -0.08 298.53159 -5.0314374 0.046526108 0.0042843939 154.31436 -13202.054 16602.09
9000 -0.09 289.63258 -5.5876729 0.0064037479 0.07394267 149.71436 -13172.278 18357.755
10000 -0.1 284.07377 -5.8774427 0.011876434 0.017658887 146.84095 -13140.895 19493.024
11000 -0.11 320.11761 -4.2791145 0.062847323 0.072604489 165.47242 -13130.428 13812.209
12000 -0.12 384.47793 -0.26899312 0.06384105 0.075858441 198.74099 -13172.063 430.97876
13000 -0.13 406.8698 -2.3913058 -0.036697295 -0.037783423 210.31561 -13200.26 8219.2883
14000 -0.14 385.84693 -3.3014902 0.059581512 -0.048723245 199.44864 -13199.524 10968.773
15000 -0.15 365.16317 -4.0204884 0.058056622 0.017209135 188.75697 -13191.173 13150.742
16000 -0.16 352.29941 -4.8141755 0.052769795 -0.039907224 182.10755 -13179.442 16004.377
17000 -0.17 333.83554 -5.4774396 0.014373592 -0.0069549415 172.56337 -13157.84 18233.403
18000 -0.18 326.9576 -6.0482935 0.022646093 0.050253999 169.00809 -13135.624 19917.978
19000 -0.19 328.65703 -5.4349022 -0.052976878 0.02948022 169.88654 -13113.032 18194.663
20000 -0.2 402.8138 -1.2165489 0.030833241 -0.037641657 208.21901 -13158.984 4077.8578
Loop time of 95.4143 on 8 procs for 20000 steps with 4000 atoms
Pair time (%) = 74.0979 (77.6591)
Neigh time (%) = 0.127079 (0.133187)
Comm time (%) = 11.7939 (12.3607)
Outpt time (%) = 2.51201 (2.63274)
Other time (%) = 6.8834 (7.21423)
Nlocal: 500 ave 507 max 495 min
Histogram: 2 1 0 0 2 1 1 0 0 1
Nghost: 2485.88 ave 2494 max 2479 min
Histogram: 1 1 2 0 1 0 0 1 0 2
Neighs: 35096 ave 36398 max 34125 min
Histogram: 2 0 2 1 0 0 0 2 0 1
FullNghs: 70196.5 ave 71230 max 69522 min
Histogram: 3 0 0 0 2 1 1 0 0 1
Total # of neighbors = 561572
Ave neighs/atom = 140.393
Neighbor list builds = 62
Dangerous builds = 0
print "All done"
All done
LAMMPS datafile
The following datafile, Al_comp.def1.txt, was also generated during this simulation.
# Fix print output for fix def1 -0.001000000088 -0.06084024339 0.005103309452 0.011634529 -0.002000000088 -0.1414344573 -0.02482512452 -0.00442782696 -0.003000000088 -0.1893439523 0.009944994037 0.01031293284 -0.004000000088 -0.2745471681 -0.01431999273 0.006837682636 -0.005000000088 -0.3417247015 -0.005247107989 -0.01437892764 -0.006000000087 -0.3715769192 0.03762760787 0.03762638331 -0.007000000087 -0.4593169714 -0.03208865692 0.002251496376 -0.008000000087 -0.5124145984 0.00431254376 0.04566519111 ...
LAMMPS dumpfile
The following is the beginning of one of the dumpfiles in cfg format that were also generated during this simulation.
Number of particles = 4000 A = 1.0 Angstrom (basic length-scale) H0(1,1) = 40.6491 A H0(1,2) = 0 A H0(1,3) = 0 A H0(2,1) = 0 A H0(2,2) = 40.6491 A H0(2,3) = 0 A H0(3,1) = 0 A H0(3,2) = 0 A H0(3,3) = 40.6491 A .NO_VELOCITY. entry_count = 8 auxiliary[0] = csym auxiliary[1] = peratom auxiliary[2] = fx auxiliary[3] = fy auxiliary[4] = fz 26.982 Al 0.0493442 0.0581882 0.00323696 1.58804 -3.34266 0.196691 -0.804728 -0.0280286 0.0987502 0.00417997 0.00238952 0.827133 -3.34273 -0.475844 -0.270217 -0.120716 0.0989802 0.0523976 0.0521907 0.363772 -3.30304 -0.309005 -0.274515 -0.299433 0.0495058 0.102418 0.0536544 0.611128 -3.30518 0.162097 -0.42069 -0.15808 0.0995051 0.102793 0.00119416 0.614289 -3.28181 -0.406727 -0.192102 0.405813 0.0473774 0.0488825 0.10204 0.581043 -3.3709 0.135458 -0.205472 -0.294232 0.0989535 0.000697083 0.0977144 0.583569 -3.30702 -0.474259 0.0845695 0.400838 ...
Post-Processing
Stress-Strain Plot
The stress-strain curve in Figure 1 can be generated using the following MATLAB script. Note that the definitions of stress and strain are negative. This is done to counteract the negative values of compressive stress and strain so that positive axes and a familiar shape are retained. The "exportfig" command saves the plot to a tiff files, but the plot can also be saved as a Mathcad figure once it appears.
%% Analyze def1.txt files
% Plot the various responses
d = dir('*.def1.txt');
for i = 1:length(d)
% Get data
fname = d(i).name;
A = importdata(fname);
strain = A.data(:,1);
stress = A.data(:,2:4);
% Generate plot
plot(strain,stress(:,1),'-or','LineWidth',2,'MarkerEdgeColor','r',...
'MarkerFaceColor','r','MarkerSize',5),hold on
plot(strain,stress(:,2),'-ob','LineWidth',2,'MarkerEdgeColor','b',...
'MarkerFaceColor','b','MarkerSize',5),hold on
plot(strain,stress(:,3),'-og','LineWidth',2,'MarkerEdgeColor','g',...
'MarkerFaceColor','g','MarkerSize',5),hold on
axis square
ylim([0 10])
set(gca,'LineWidth',2,'FontSize',24,'FontWeight','normal','FontName','Times')
set(get(gca,'XLabel'),'String','Strain','FontSize',32,'FontWeight','bold','FontName','Times')
set(get(gca,'YLabel'),'String','Stress (GPa)','FontSize',32,'FontWeight','bold','FontName','Times')
set(gcf,'Position',[1 1 round(1000) round(1000)])
% Export figure to tif file
exportfig(gcf,strrep(fname,'.def1.txt','.tif'),'Format','tiff',...
'Color','rgb','Resolution',300)
close(1)
end
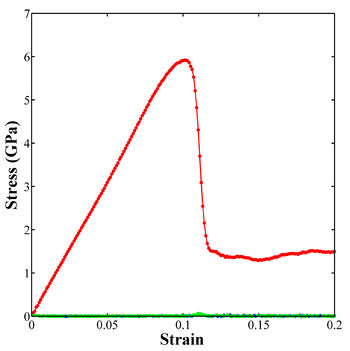
Figure 1. Stress-strain curve for uniaxial compressive loading of single crystal aluminum in the <100> loading direction.
Deformation Movie
This assumes that you already have AtomEye and ImageJ downloaded.- Visualize the dumpfile in AtomEye by typing the following command, "/A dump.tensile_0.cfg" (UNIX).
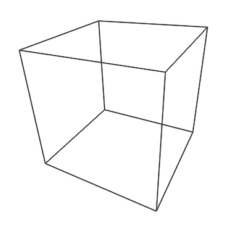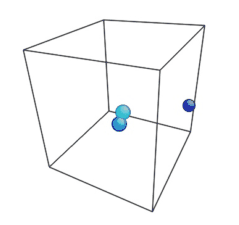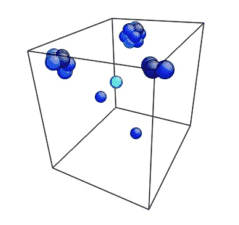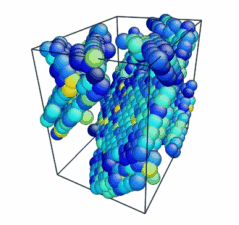
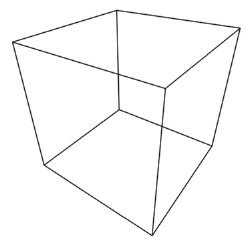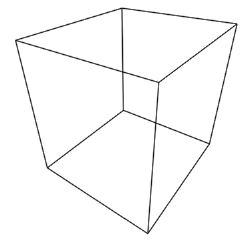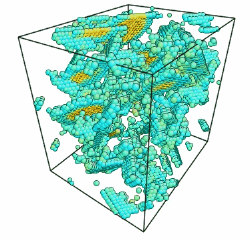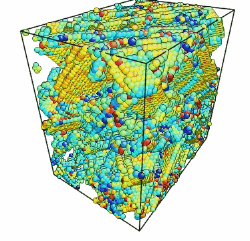
- Use the AtomEye options to select how you want to visualize deformation. In this example, the centrosymmetry parameter was used to show only atoms in a non-centrosymmetric environment (see Fig. 2).
- Use Alt+0 to activate centrosymmetric (csym) view.
- Adjust threshold, or set of atoms to view, by using Shift+T. This will allow creation of a set for the current parameter (in this case, csym). Please note that you need to adjust both lower and higher thresholds unless the atoms from following images that exceeds maximum value for the first one will be not shown. You can make it 5 or 10.
- Make atoms with values outside of the threshold invisible by using Ctrl+A.
- Press 'y' within AtomEye to produce an animation script.
- The folder "Jpg" now contains snapshots of all dumpfiles.
- Open ImageJ
- Drag the folder Jpg into ImageJ
- Select "Convert to RGB" to keep the color from the AtomEye images.
- Choose "yes" to load a stack.
- Adjust the size as needed (Image/Adjust/Size)
- Adjust frame rate as desired (Image/Stacks/Animation Options)
- Save as Animated Gif file
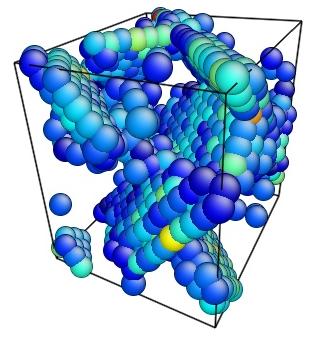
Figure 2. Image of nucleated dislocation near peak stress.
Movie showing compressive deformation of single crystal aluminum loaded in the <100> direction at a strain rate of 1010 s-1 and a temperature of 300 K. Only atoms in non-centrosymmetric environment are shown.
Deformation Movie using OVITO
In order for LAMMPS to output compatible data, one must set up an appropriate dump file with the desired data in the LAMMPS input script. Near the end of the LAMMPS script, use the 'dump' command in order to output desired data to a 'custom' file type for use in Ovito. The following lines create a dumpfile for every atom in the simulation every 250 timesteps, and each file is named according to its associated timestep. Then, the file is specified to show, for each atom, the atom ID, atom type, scaled atom coordinates, previously computed centrosymmetry and potential energy variables, and forces upon each atom.
# Dump to cfg for Ovito post processing dump 1 all custom 250 dump.comp.* id type xs ys zs c_csym c_peratom fx fy fz
Now to make a movie using OVITO:
- Open Ovito.
- Select "File/Import," one of the dumpfiles you wish to animate, and "Open."
- Select "Use following wild-card name to load multiple files."
- You must tell Ovito what each column of data in the dumpfile is. Often, Ovito can do this for you if you click "Auto-assign columns."
- Use the Modifier List to add the desired modifications to your images and future animation. For this example, the "Common Neighbor Analysis" modifier was used to visualize dislocations.
- Test the animation by using the animation tools below the graphics.
- Click the "Render" tab.
- Select "Complete animation" and choose an output filename in "Output Filename." Save to a new directory for use later in ImageJ. Be sure to include an appropriate extension, such as ".bmp."
- Select the view that you would like to animate and click "Render Active Viewport" above "Render settings."
- Now you should have an image for every frame of your simulation.
Cell Size Comparison
In order to test the difference that the number of atoms can have on a simulation, the above script was run with 32,000 and 108,000 atoms in addition 4,000 atoms. Editing the values (currently "10") in the following line in the input script will change the size of the simulation cell and the number of atoms used in the simulation. Values of "20" will result in 32,000 atoms, and values of "30" will result in 108,000 atoms.
region whole block 0 10 0 10 0 10
Shown below are movies of the 4,000, 32,000, and 108,000 atom simulations, which show only atoms in a non-centrosymmetric environment. As one might expect, more slip planes become visible as the atom count of the simulation increases.
Movie showing compressive deformation of a 4,000 atom single crystal of aluminum loaded in the <100> direction. Only atoms in non-centrosymmetric environment are shown.
Movie showing compressive deformation of a 32,000 atom single crystal of aluminum loaded in the <100> direction. Only atoms in non- centrosymmetric environment are shown.
Movie showing compressive deformation of a 108,000 atom single crystal of aluminum loaded in the <100> direction. Only atoms in non-centrosymmetric environment are shown.
Temperature Comparison
The temperature of the simulations also makes a difference in the outputs. To show this difference, the above simulations (previously at 300 K) were run at 10 K. In order to change the temperature of the simulation, three lines in the input script must be edited. Two lie under the 'Equilibraion' section while the third lies under 'Deformation.'
The velocity and fix commands shown below contain temperature data. The velocity command specifies the thermal velocity of the system while the fix command specifies the desired temperatures at the beginning and end of the simulation. In order to run the simulation at 10 K instead of 300 K, change the three '300' values to '10' in the velocity and fix command lines.
# EQUILIBRATION reset_timestep 0 timestep 0.001 velocity all create 300 12345 mom yes rot no fix 1 all npt temp 300 300 1 iso 0 0 1 drag 1
The temperature values in the fix command under 'Deformation' also needs to be changed to '10' instead of '300.' The values are being changed again because between 'Equilibration' and 'Deformation' the fix ID 1 was unfixed. Here fix 1 is being redefined.
# DEFORMATION reset_timestep 0 fix 1 all npt temp 300 300 1 y 0 0 1 z 0 0 1 drag 1
All three simulation cell sizes were run at 10 K. Below are the movies from the simulations. Once again, only atoms in a non-centrosymmetric environment are viewable. The difference between the 300 K and 10 K simulations is that there is less non-centrosymmetry induced by thermal velocity. At 10 K, many fewer atoms are seen before slip occurs, and the slip planes are more cleary visible and absent of "noise" created by atoms that are non-centrosymmetric solely due to thermal activity.
Movie showing compressive deformation of a 4,000 atom single crystal of aluminum at 10 K loaded in the <100> direction. Only atoms in non-centrosymmetric environment are shown.
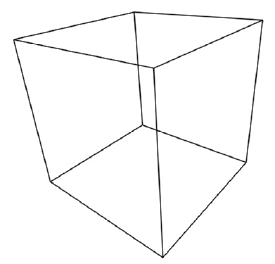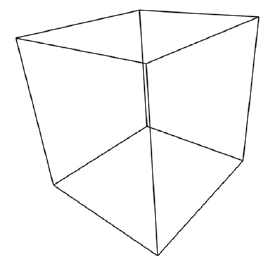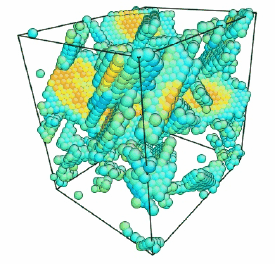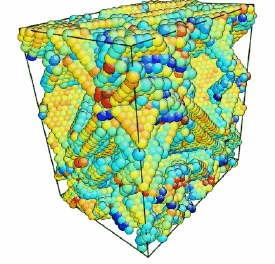
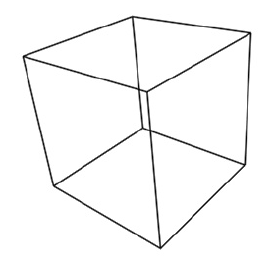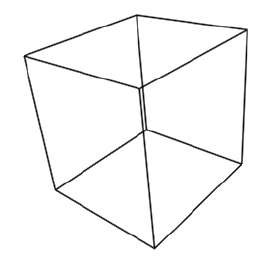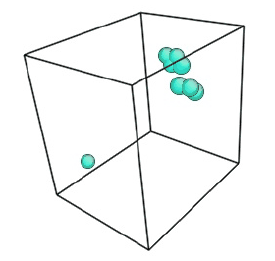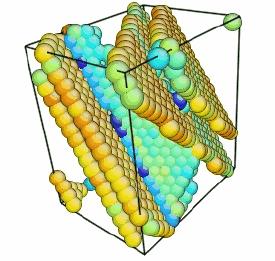
Movie showing compressive deformation of a 32,000 atom single crystal of aluminum at 10 K loaded in the <100> direction. Only atoms in non-centrosymmetric environment are shown.
Movie showing compressive deformation of a 108,000 atom single crystal of aluminum at 10 Kloaded in the <100> direction. Only atoms in non-centrosymmetric environment are shown.
Questions and Answers?
Q1: I noticed that on the stress-strain curve in the Figure 1, following the peak stress (dislocation nucleation), the stresses are approximately 2 GPa in the strain range 0.1-0.2. However, in the log file and output files for the input script above, the stresses range from 2-6 GPa. Why?
A1: Aha! We threw a curveball at you. The plot is actually for a much larger simulation cell. The 4000-atom simulation cell from the above input script is relatively small. Hence, after dislocation nucleation, further plastic deformation becomes very difficult and you will actually see a second curve. For much larger cell sizes (and more atoms), this flow stress following nucleation should begin to look more like the stress-strain curve in Figure 1. This observation also brings up an important point - the behavior and properties calculated using molecular dynamics can sometimes be heavily influenced by the size of the simulation cell (and the strain rate and the potential and the temperature and the boundary conditions). I relate this size dependence in MD simulations to a mesh sensitivity study in finite element models - you should keep increasing the simulation cell size to understand the potential error in your calculations. If you can obtain some degree of convergence in properties or behavior (mechanism), this is the ideal situation.
Acknowledgments
The authors would like to acknowledge funding for this work through the Department of Energy.
References
- ↑ S. Plimpton, "Fast Parallel Algorithms for Short-Range Molecular Dynamics," J. Comp. Phys., 117, 1-19 (1995).
- ↑ Spearot, D.E., Tschopp, M.A., Jacob, K.I., McDowell, D.L., "Tensile strength of <100> and <110> tilt bicrystal copper interfaces," Acta Materialia 55 (2007) p. 705-714 (http://dx.doi.org/10.1016/j.actamat.2006.08.060).
- ↑ Tschopp, M.A., Spearot, D.E., McDowell, D.L., "Atomistic simulations of homogeneous dislocation nucleation in single crystal copper," Modelling and Simulation in Materials Science and Engineering 15 (2007) 693-709 ( http://dx.doi.org/10.1088/0965-0393/15/7/001).
- ↑ Tschopp, M.A., McDowell, D.L., "Influence of single crystal orientation on homogeneous dislocation nucleation under uniaxial loading," Journal of Mechanics and Physics of Solids 56 (2008) 1806-1830. (http://dx.doi.org/10.1016/j.jmps.2007.11.012).
- ↑ Y. Mishin, D. Farkas, M.J. Mehl, and D.A. Papaconstantopoulos, "Interatomic potentials for monoatomic metals from experimental data and ab initio calculations," Phys. Rev. B 59, 3393 (1999).
- ↑ J. Li, "AtomEye: an efficient atomistic configuration viewer," Modelling Simul. Mater. Sci. Eng. 11 (2003) 173.