
Integrated Computational Materials Engineering (ICME)
LAMMPS Tutorial 1
Abstract
This is a quick tutorial to running a LAMMPS simulation on a Windows machine. For this simple example, the molecular simulation calculates the equilibrium lattice constant and corresponding cohesive energy for aluminum.
Author(s): Mark A. Tschopp
The general workflow for running molecular dynamics simulations using LAMMPS is illustrated in the figure below:
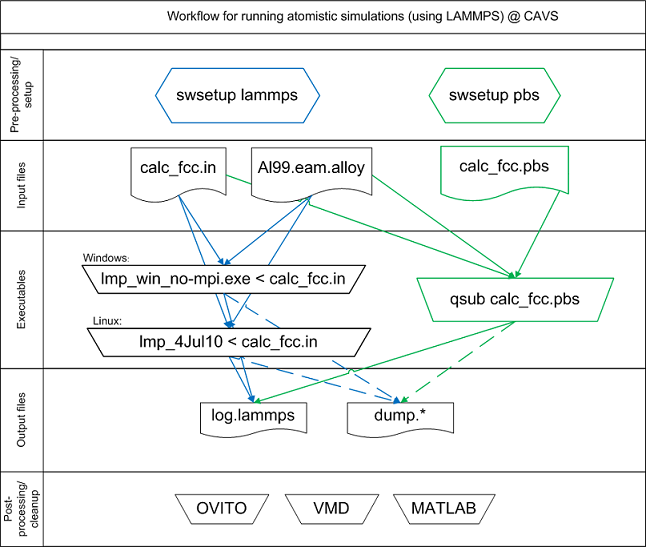
Figure 1. The face-centered cubic lattice structure.
Download LAMMPS
Follow these steps to download the LAMMPS Windows Executable:- Goto the LAMMPS download webpage, click here.
- Click on LAMMPS Windows serial executable.
- Click the "download now" button.
If you wish to run LAMMPS in a Unix shell, you must download a version on download webpage and compile and executable.
Voila! Easy as that.
Download a potential
Follow these steps to download a potential.- Goto the NIST Interatomic Potential webpage, click here.
- Click on Al for an aluminum single element potential.
- Click on whichever potential you like. For this example, use the Mishin et al. potential, Al99.eam.alloy.
Save this in the same directory as the LAMMPS executable.
Download an input file
This input script was run using the Aug 2015 version of LAMMPS. Changes in some commands may require revision of the input script. Copy the text below and paste it into a text file, 'calc_fcc.in'. Use the 'Paste Special' command with 'Unformatted Text'.
# Find minimum energy fcc configuration
# Mark Tschopp, 2010
# ---------- Initialize Simulation ---------------------
clear
units metal
dimension 3
boundary p p p
atom_style atomic
atom_modify map array
# ---------- Create Atoms ---------------------
lattice fcc 4
region box block 0 1 0 1 0 1 units lattice
create_box 1 box
lattice fcc 4 orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
create_atoms 1 box
replicate 1 1 1
# ---------- Define Interatomic Potential ---------------------
pair_style eam/alloy
pair_coeff * * Al99.eam.alloy Al
neighbor 2.0 bin
neigh_modify delay 10 check yes
# ---------- Define Settings ---------------------
compute eng all pe/atom
compute eatoms all reduce sum c_eng
# ---------- Run Minimization ---------------------
reset_timestep 0
fix 1 all box/relax iso 0.0 vmax 0.001
thermo 10
thermo_style custom step pe lx ly lz press pxx pyy pzz c_eatoms
min_style cg
minimize 1e-25 1e-25 5000 10000
variable natoms equal "count(all)"
variable teng equal "c_eatoms"
variable length equal "lx"
variable ecoh equal "v_teng/v_natoms"
print "Total energy (eV) = ${teng};"
print "Number of atoms = ${natoms};"
print "Lattice constant (Angstoms) = ${length};"
print "Cohesive energy (eV) = ${ecoh};"
print "All done!"
Run this using LAMMPS in Windows, Method 1
Follow these steps if running LAMMPS in Windows:- Click on the Start button on the toolbar
- Click on Run...
- Enter 'cmd' and hit OK
- In new window, change to the directory that contains the LAMMPS executable (lmp_win_no-mpi.exe), the input script (calc_fcc.in), and the potential file (Al99.eam.alloy). For example, if these are on the desktop, type 'cd h:/desktop' to change to the desktop.
- Type 'lmp_win_no-mpi < calc_fcc.in'
- If the simulation completes, then it will type "All done!"
Run this using LAMMPS in Windows, Method 2
An alternative way to run this in Windows is to develop a *.bat file, which is a Windows executable file. For instance, create a new text file and change the extension from .txt to .bat. By double clicking on a *.bat file, you will execute the lines of scripts within the file. Now, don't double click to open, but rather right click and click on edit. Then enter the following lines:
lmp_win_no-mpi.exe < calc_fcc.in echo All done! echo Press a key to end pause > nul
Try it by dropping it in the same folder as the LAMMPS executable, input script, and potential file and double clicking... voila!
Run this using LAMMPS in UNIX
If in Unix, simply type 'LAMMPS executable < input file.' If you wish to use multiple processors, use the mpirun command. For example, 'mpirun -np 8 LAMMPS executable < input file' runs the simulation on 8 processors.
The end of the logfile/screen output should look like this:
Total energy (eV) = -13.43999995; Number of atoms = 4; Lattice constant (Angstoms) = 4.05; Cohesive energy (eV) = -3.359999988; All done!
What it does
In this section, we will break down what LAMMPS is doing for each step. The easy way to do this on your own is to consult the LAMMPS manual for each command or go to the Internet LAMMPS manual, i.e., at lammps.sandia.gov.
# Find minimum energy fcc configuration # Mark Tschopp, 2010
The '#' denotes a comment. Hence LAMMPS does not do anything for these lines.
# ---------- Initialize Simulation --------------------- clear units metal dimension 3 boundary p p p atom_style atomic atom_modify map array
This section initializes the simulations. The 'clear' command clears all memory. The 'units' command specifies the units that will be used for the remainder of the simulation; 'metal' uses Angstroms and eV, among other units. The 'dimension 3' command specifies a 3-dimensional simulation cell will be used (2-D is also an option). The 'boundary p p p' specifies periodic boundaries in the x-, y-, and z-direction.
# ---------- Create Atoms --------------------- lattice fcc 4 region box block 0 1 0 1 0 1 units lattice create_box 1 box lattice fcc 4 orient x 1 0 0 orient y 0 1 0 orient z 0 0 1 create_atoms 1 box replicate 1 1 1
The 'lattice' command specifies what type of lattice is used (fcc, bcc, hcp, etc.) and the number following this specifies the lattice constant. The 'region' command specifies the simulation cell. Here, we have used lattice units and specified that the simulation cell box is to be 1 lattice unit in each direction. The 'create_box' command following this will use the parameters outlined in the 'region' command to actually create the box. The 'replicate' command can be used to replicate the periodic cell in each direction.
# ---------- Define Interatomic Potential --------------------- pair_style eam/alloy pair_coeff * * Al99.eam.alloy Al neighbor 2.0 bin neigh_modify delay 10 check yes
The 'pair_style' command specifies what kind of interatomic potential will be used, while the 'pair_coeff' specifies the file that the pair potential coefficients are stored in. HINT: the potential extension can sometimes give a clue as to which format is being used.
# ---------- Define Settings --------------------- compute eng all pe/atom compute eatoms all reduce sum c_eng
Here, two computes are defined. In the first 'compute' command, a variable named 'eng' is defined to store the potential energy for each atom. In the second 'compute' command, a variable named 'eatoms' is defined to store the sum of all 'eng' values
# ---------- Run Minimization --------------------- reset_timestep 0 fix 1 all box/relax iso 0.0 vmax 0.001 thermo 10 thermo_style custom step pe lx ly lz press pxx pyy pzz c_eatoms min_style cg minimize 1e-25 1e-25 5000 10000
The 'reset_timestep' does just that. The 'fix' command uses the 'box/relax' setting, whereby all directions ('iso') are relaxed to 0.0 Pa pressure for all atoms ('all'). The 'thermo' specifies the output during minimization. The 'thermo_style' specifies what type of output is shown to screen. The 'min_style' specifies that conjugate gradient will be used for minimization and the 'minimize' starts the minimization, i.e., the simulation cell boundaries are relaxed from the specified lattice constant (4 Angstroms) to the actual lattice constant (4.05 Angstroms).
variable natoms equal "count(all)" variable teng equal "c_eatoms" variable length equal "lx" variable ecoh equal "v_teng/v_natoms"
This section defines some variables, such as 'natoms' for the number of atoms, 'teng' for the total potential energy, 'length' for the length of the simulation cell, and 'ecoh' for the cohesive energy of Al.
print "Total energy (eV) = ${teng};"
print "Number of atoms = ${natoms};"
print "Lattice constant (Angstoms) = ${length};"
print "Cohesive energy (eV) = ${ecoh};"
print "All done!"
This section prints these values to screen and to the logfile. The '${}' is used to insert the variables defined earlier.
Questions and Answers
Q1: I typed in the above line and nothing happened.
A1: Make sure that you are in the same directory as the LAMMPS executable. Make sure that you are typing in the correct filename.
Q2: I keep getting an error with the potential, i.e., potential file not found.
A2: First, check that the potential file is in the directory that you are running out of. Although, you can insert paths, if you want to store the potentials in another directory. Second, make sure that the potential file name is the same as that given in the input script. For instance, Windows has the habit of saving text files (like the potential files) with .txt extensions. If you happen to be running from a Windows operating system, I would change the settings so that you can see the extensions of the files as well.