Integrated Computational Materials Engineering (ICME)
LAMMPS reactive deformation of a single polyethylene chain
Abstract
In this tutorial, molecular dynamics simulation in LAMMPS is used to show the deformation of a single polymer chain using the Modified Embedded Atom Method (MEAM) potential for hydrocarbons (C/H). This example script first performs an energy minimization and equilibration on the polymer chain. Then, the chain is deformed by fixing one end of the chain to an imaginary wall while applying a velocity to the other end of the chain. With the utilization of the reactive MEAM potential we can directly observe bond rupture caused by deformation. Using OVITO, the deformation and chain rupture can be visualized.
Author(s): Andrew Bowman, Sungkwang Mun.
Corresponding Author: Andrew Bowman
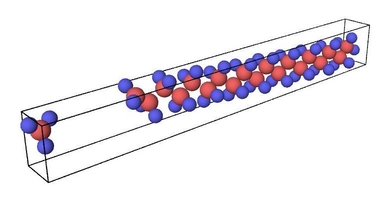
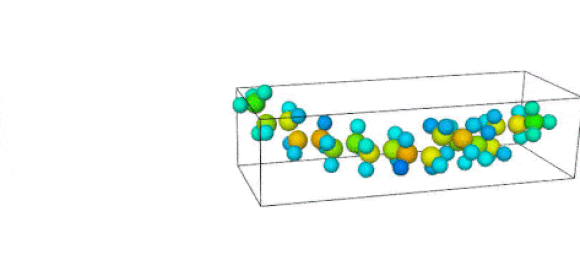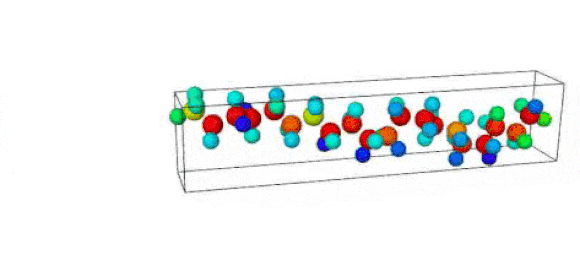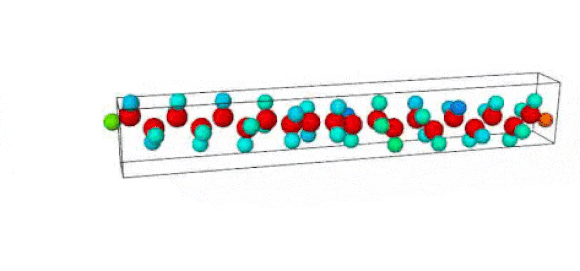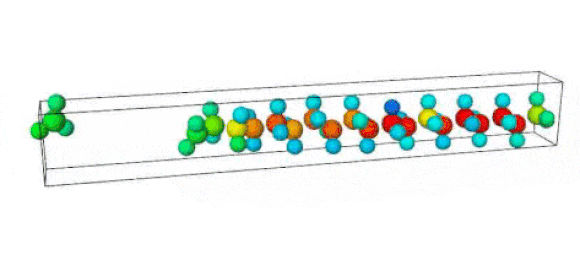
Figure 1. Ruptured polymer chain visualized in OVITO.
Input File
The polymer chain of 62 atoms (20 carbon, 42 hydrogen) was prepared in Accelrys Materials Studio. For the MEAM potential, each atom, including hydrogen atoms, must be explicitly represented in the input file. Additional open source polymer creation tools are available or a custom code can be used. For this simulation the polymer was built along the y-direction as indicated in Fig. 2
The input data file is shown below:
# nc1cl10
62 atoms
-2.377300 5.360300 xlo xhi
0.590000 21.363700 ylo yhi
-2.163700 3.024900 zlo zhi
2 atom types
Masses
1 12.0111
2 1.0079
Atoms
1 1 1.000000 1.540000 0.000000
2 1 2.456100 2.041400 0.000000
3 1 1.538500 4.082700 -1.130600
4 1 2.456100 3.581400 0.000000
5 1 1.538500 5.622700 -1.130600
6 1 0.370100 6.124100 -0.261600
7 1 0.370100 7.664100 -0.261600
8 1 -0.875500 8.165500 0.492500
9 1 -0.875500 9.705500 0.492500
10 1 -1.359100 10.206800 -0.881000
11 1 -1.359100 11.746800 -0.881000
12 1 0.095700 12.248200 -0.944400
13 1 0.095700 13.788200 -0.944400
14 1 1.468100 14.289600 -0.458000
15 1 2.898200 16.331000 -0.183900
16 1 1.468100 15.829600 -0.458000
17 1 2.898200 17.871000 -0.183900
18 1 3.201500 18.372300 1.240300
19 1 4.452800 20.413700 1.985000
20 1 3.201500 19.912300 1.240300
21 2 1.000000 0.590000 0.000000
22 2 0.480000 1.898100 0.900700
23 2 0.480000 1.898100 -0.900700
24 2 2.964000 1.666300 -0.900700
25 2 2.964000 1.666300 0.900700
26 2 1.910100 3.724600 -2.102000
27 2 0.511400 3.724600 -0.966700
28 2 2.076800 3.956500 0.962000
29 2 3.475500 3.956500 -0.173300
30 2 1.418200 5.980900 -2.163700
31 2 2.493300 5.980900 -0.718200
32 2 0.500100 5.749000 0.764300
33 2 -0.575000 5.749000 -0.681200
34 2 0.348500 8.022200 -1.301400
35 2 1.281400 8.022200 0.239700
36 2 -0.843600 7.790400 1.526100
37 2 -1.776500 7.790400 -0.015000
38 2 0.146800 10.063600 0.683900
39 2 -1.552500 10.063600 1.282100
40 2 -2.377300 9.831700 -1.061000
41 2 -0.678100 9.831700 -1.659200
42 2 -1.839400 12.105000 0.041600
43 2 -1.917800 12.105000 -1.758200
44 2 0.563900 11.873100 -1.866400
45 2 0.642300 11.873100 -0.066600
46 2 -0.695400 14.146300 -0.269100
47 2 -0.093600 14.146300 -1.967100
48 2 2.247700 13.914500 -1.137300
49 2 1.646000 13.914500 0.560700
50 2 3.239400 15.972800 0.798600
51 2 3.578500 15.972800 -0.970600
52 2 1.138800 16.204700 -1.438200
53 2 0.799700 16.204700 0.331100
54 2 1.908900 18.229100 -0.504900
55 2 3.670800 18.229100 -0.880100
56 2 4.188300 17.997200 1.549400
57 2 2.426400 17.997200 1.924700
58 2 4.452800 21.363700 1.985000
59 2 4.439000 20.055600 3.024900
60 2 5.360300 20.055600 1.476900
61 2 3.225700 20.287400 0.206500
62 2 2.304400 20.287400 1.754500
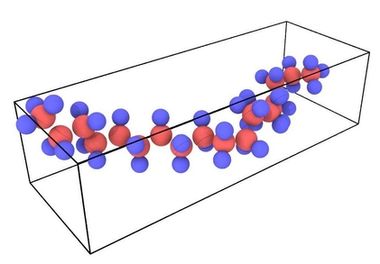
Figure 2. Undeformed polymer chain visualized in OVITO.
LAMMPS Script
Below is the script used for the actual simulation. The Aug 2015 version of LAMMPS was used. The MEAM potential for saturated hydrocarbons was used for this simulation. The required library.meam and CH.meam parameters can be found here. Minimization and equilibration were performed first, then atom groups were defined to allow for chain stretching at a constant velocity. The compute " stress/atom" was used to visualize the stress distribution along the chain. This input script was run using the February 2014 version of LAMMPS. Changes in some commands in more recent versions may require revision of the input script. This script runs the simulation with the previously discussed input file.
#Initialization
units metal
boundary s s s
atom_style atomic
variable filename string nc1cl10
read_data ${filename}.dat
# Meam Potential for Saturated Hydrocarbons Information
pair_style meam
pair_coeff * * library.meam C H CH.meam C H
##################################################################################
#Minimization
minimize 0.0 1.0e-8 100000 100000
# Equilibration
reset_timestep 0
velocity all create 0.0 1231 dist gaussian
fix 1 all nve
fix 2 all langevin 0 0 0.001 904297
thermo_style custom step etotal press vol temp density pxx pyy pzz
thermo 100
timestep 0.0003
run 20000
unfix 1
unfix 2
######################################################################
# Deformation
run 0
reset_timestep 0
#Define Variables
variable yl equal "ylo+1"
variable yh equal "yhi-1"
variable yh2 equal "yhi*3"
variable y_step equal "yhi/2"
variable p3 equal "ly"
variable L0 equal ${p3}
variable strain equal "(ly - v_L0)/v_L0"
variable p1 equal v_strain
variable p2 equal "-pyy/10"
# define groups
region 1 block INF INF INF ${yl} INF INF
group wall region 1
region 2 block INF INF ${yh} INF INF INF
group pull region 2
group boundary union wall pull
group mobile subtract all wall pull
# computes
compute peratom all stress/atom NULL pair
# initial velocities
velocity mobile create 0.01 887723
velocity pull set 0.0 .5 0.0
velocity mobile ramp vy 0 ${y_step} y 0 ${yh2} sum yes
# fixes
fix f1 all nve
fix ftemp all langevin 25 25 0.05 904297
fix f2 boundary setforce NULL 0.0 0.0
# Dump
dump 100 all custom 1000 stress_per_atom.txt id type x y z c_peratom[1] c_peratom[2] c_peratom[3] c_peratom[4] c_peratom[5] c_peratom[6]
fix def1 all print 100 "${p1} ${p2}" file Stress_strain.def1.txt screen no
thermo_style custom step temp pyy lx ly lz v_strain
thermo 1000
timestep 0.0005
run 50000
print "All Done"
LAMMPS Logfile
Here is the logfile produced by LAMMPS during the simulation.
LAMMPS (1 Feb 2014)
#Initialization
units metal
boundary s s s
atom_style atomic
variable filename string nc1cl10
read_data ${filename}.dat
read_data nc1cl10.dat
orthogonal box = (-2.3773 0.59 -2.1637) to (5.3603 21.3637 3.0249)
2 by 5 by 1 MPI processor grid
reading atoms ...
62 atoms
# Meam Potential for Saturated Hydrocarbons Information
pair_style meam
pair_coeff * * library.meam C H CH.meam C H
dump d1 all movie 100 movie.mp4 type type size 640 480 zoom 2.5
##################################################################################
#Minimization
minimize 0.0 1.0e-8 100000 100000
WARNING: Resetting reneighboring criteria during minimization (../min.cpp:173)
Memory usage per processor = 5.7362 Mbytes
Step Temp E_pair E_mol TotEng Press Volume
0 0 -256.88642 0 -256.88642 16039.246 834.50871
2275 0 -258.98482 0 -258.98482 -15.964463 880.60809
Loop time of 9.09964 on 10 procs for 2275 steps with 62 atoms
Minimization stats:
Stopping criterion = linesearch alpha is zero
Energy initial, next-to-last, final =
-256.886423993 -258.984817441 -258.984817441
Force two-norm initial, final = 17.9272 0.00267172
Force max component initial, final = 8.69935 0.00144996
Final line search alpha, max atom move = 0.00195312 2.83195e-06
Iterations, force evaluations = 2275 18266
Pair time (%) = 5.0886 (55.9209)
Neigh time (%) = 3.75032e-05 (0.00041214)
Comm time (%) = 1.83001 (20.1108)
Outpt time (%) = 0.740981 (8.14297)
Other time (%) = 1.44001 (15.8249)
Nlocal: 6.2 ave 13 max 0 min
Histogram: 2 0 1 2 0 0 3 0 0 2
Nghost: 35.4 ave 53 max 18 min
Histogram: 1 1 1 1 0 2 2 1 0 1
Neighs: 92.8 ave 190 max 0 min
Histogram: 2 0 1 0 2 2 0 1 0 2
FullNghs: 185.6 ave 406 max 0 min
Histogram: 2 0 2 1 1 0 1 1 1 1
Total # of neighbors = 1856
Ave neighs/atom = 29.9355
Neighbor list builds = 1
Dangerous builds = 0
# Equilibration
reset_timestep 0
velocity all create 0.0 1231 dist gaussian
fix 1 all nve
fix 2 all langevin 0 0 0.001 904297
thermo_style custom step etotal press vol temp density pxx pyy pzz
thermo 1000
timestep 0.0003
run 20000
Memory usage per processor = 5.04914 Mbytes
Step TotEng Press Volume Temp Density Pxx Pyy Pzz
0 -258.98482 -16.335355 860.61399 0 0.54518234 4.2240815 -52.460361 -0.76978563
1000 -258.98482 -18.467099 860.61399 2.6373086e-07 0.54518234 4.279785 -58.922537 -0.75854527
2000 -258.98482 -18.408935 860.61399 2.57724e-07 0.54518234 4.3413454 -58.764647 -0.80350341
3000 -258.98482 -18.351508 860.61399 2.5287401e-07 0.54518234 4.3926884 -58.599195 -0.84801847
4000 -258.98482 -18.296634 860.61399 2.4862843e-07 0.54518234 4.4353853 -58.435041 -0.89024532
5000 -258.98482 -18.244933 860.61399 2.4477753e-07 0.54518234 4.4695285 -58.274125 -0.93020235
6000 -258.98482 -18.196254 860.61399 2.4121733e-07 0.54518234 4.4957193 -58.11642 -0.9680618
7000 -258.98483 -18.150042 860.61399 2.3788626e-07 0.54518234 4.514807 -57.961022 -1.0039105
8000 -258.98483 -18.105594 860.61399 2.3474327e-07 0.54518234 4.5277382 -57.80672 -1.0377988
9000 -258.98483 -18.06221 860.61399 2.3175872e-07 0.54518234 4.5354502 -57.652305 -1.0697762
10000 -258.98483 -18.01927 860.61399 2.2891003e-07 0.54518234 4.538808 -57.496715 -1.0999023
11000 -258.98483 -17.976252 860.61399 2.2617938e-07 0.54518234 4.5385756 -57.339085 -1.1282465
12000 -258.98483 -17.93274 860.61399 2.2355234e-07 0.54518234 4.5354099 -57.178746 -1.1548854
13000 -258.98483 -17.888417 860.61399 2.2101696e-07 0.54518234 4.5298616 -57.015212 -1.1798998
14000 -258.98483 -17.843046 860.61399 2.1856326e-07 0.54518234 4.5223879 -56.848154 -1.2033727
15000 -258.98483 -17.796462 860.61399 2.1618277e-07 0.54518234 4.513367 -56.677366 -1.2253871
16000 -258.98484 -17.748553 860.61399 2.1386829e-07 0.54518234 4.5031093 -56.502744 -1.2460252
17000 -258.98484 -17.699254 860.61399 2.1161362e-07 0.54518234 4.4918689 -56.324264 -1.2653669
18000 -258.98484 -17.648533 860.61399 2.094134e-07 0.54518234 4.4798528 -56.141963 -1.2834899
19000 -258.98484 -17.596388 860.61399 2.07263e-07 0.54518234 4.46723 -55.955925 -1.3004684
20000 -258.98484 -17.542835 860.61399 2.0515839e-07 0.54518234 4.4541384 -55.766269 -1.3163738
Loop time of 21.1757 on 10 procs for 20000 steps with 62 atoms
Pair time (%) = 5.64706 (26.6677)
Neigh time (%) = 0 (0)
Comm time (%) = 2.03111 (9.59171)
Outpt time (%) = 7.50698 (35.4509)
Other time (%) = 5.99055 (28.2897)
Nlocal: 6.2 ave 13 max 0 min
Histogram: 2 0 1 2 0 0 3 0 0 2
Nghost: 35.4 ave 53 max 19 min
Histogram: 1 1 1 1 0 2 2 1 0 1
Neighs: 93 ave 192 max 0 min
Histogram: 2 0 1 1 1 2 0 1 0 2
FullNghs: 186 ave 406 max 0 min
Histogram: 2 0 2 1 1 0 0 3 0 1
Total # of neighbors = 1860
Ave neighs/atom = 30
Neighbor list builds = 0
Dangerous builds = 0
unfix 1
unfix 2
######################################################################
# Deformation
run 0.
WARNING: No fixes defined, atoms won't move (../verlet.cpp:54)
Memory usage per processor = 5.04914 Mbytes
Step TotEng Press Volume Temp Density Pxx Pyy Pzz
20000 -258.98484 -17.55251 860.1396 2.0515839e-07 0.54548301 4.4565949 -55.797026 -1.3170998
Loop time of 1.80244e-05 on 10 procs for 0 steps with 62 atoms
Pair time (%) = 0 (0)
Neigh time (%) = 0 (0)
Comm time (%) = 0 (0)
Outpt time (%) = 0 (0)
Other time (%) = 1.80244e-05 (100)
Nlocal: 6.2 ave 13 max 0 min
Histogram: 2 0 1 2 0 0 3 0 0 2
Nghost: 35.4 ave 53 max 19 min
Histogram: 1 1 1 1 0 2 2 1 0 1
Neighs: 93 ave 192 max 0 min
Histogram: 2 0 1 1 1 2 0 1 0 2
FullNghs: 186 ave 406 max 0 min
Histogram: 2 0 2 1 1 0 0 3 0 1
Total # of neighbors = 1860
Ave neighs/atom = 30
Neighbor list builds = 0
Dangerous builds = 0
reset_timestep 0
#Define Variables
variable yl equal "ylo+1"
variable yh equal "yhi-1"
variable yh2 equal "yhi*3"
variable y_step equal "yhi/2"
variable p3 equal "ly"
variable L0 equal ${p3}
variable L0 equal 20.774388695406
variable strain equal "(ly - v_L0)/v_L0"
variable p1 equal v_strain
variable p2 equal "-pyy/10"
# define groups
region 1 block INF INF INF ${yl} INF INF
region 1 block INF INF INF 1.61162482988567 INF INF
group wall region 1
1 atoms in group wall
region 2 block INF INF ${yh} INF INF INF
region 2 block INF INF 20.3860135252917 INF INF INF
group pull region 2
1 atoms in group pull
group boundary union wall pull
2 atoms in group boundary
group mobile subtract all wall pull
60 atoms in group mobile
# computes
compute peratom all stress/atom pair
# initial velocities
velocity mobile create 0.01 887723
velocity pull set 0.0 .5 0.0
velocity mobile ramp vy 0 ${y_step} y 0 ${yh2} sum yes
velocity mobile ramp vy 0 10.6930067626458 y 0 ${yh2} sum yes
velocity mobile ramp vy 0 10.6930067626458 y 0 64.1580405758751 sum yes
# fixes
fix f1 all nve
fix ftemp all langevin 25 25 0.05 904297
fix f2 boundary setforce NULL 0.0 0.0
# Dump
dump 100 all custom 1000 stress_per_atom.txt id type x y z c_peratom[1] c_peratom[2] c_peratom[3] c_peratom[4] c_peratom[5] c_peratom[6]
fix def1 all print 100 "${p1} ${p2}" file PE2_ss_nc10_cl1000.def1.txt screen no
thermo_style custom step temp pyy lx ly lz v_strain
thermo 10000
timestep 0.0005
run 50000
Memory usage per processor = 6.66095 Mbytes
Step Temp Pyy Lx Ly Lz strain
0 7.8956181 175.93613 7.6947242 20.774389 5.3808097 1.3681129e-15
10000 24.371339 -9630.1918 6.3750014 22.806131 5.8410614 0.097800348
20000 27.667952 -15903.107 5.0358511 25.508789 5.5483935 0.22789599
30000 26.446686 -151820.66 2.8828797 28.219945 3.9085843 0.35840073
40000 25.713185 -224272.78 2.5381237 30.770621 4.4009193 0.4811806
50000 38.778302 -90892.535 2.8497228 33.235559 3.9395652 0.59983331
Loop time of 52.1821 on 10 procs for 50000 steps with 62 atoms
Pair time (%) = 11.5983 (22.2266)
Neigh time (%) = 0.000626159 (0.00119995)
Comm time (%) = 3.42308 (6.55987)
Outpt time (%) = 20.3336 (38.9666)
Other time (%) = 16.8265 (32.2458)
Nlocal: 6.2 ave 10 max 1 min
Histogram: 1 0 1 0 1 2 2 1 1 1
Nghost: 27.6 ave 40 max 16 min
Histogram: 2 0 2 1 0 1 0 2 1 1
Neighs: 71.1 ave 132 max 6 min
Histogram: 2 0 0 1 1 2 2 1 0 1
FullNghs: 142.2 ave 279 max 6 min
Histogram: 2 0 0 0 4 0 1 2 0 1
Total # of neighbors = 1422
Ave neighs/atom = 22.9355
Neighbor list builds = 26
Dangerous builds = 0
print "All Done"
All Done
LAMMPS dump files
The following dump files were produced during the simulation and can be used to make a movie. The stress-strain data was also dumped and can be used to make a stress-strain curve.
# Dump
dump 100 all custom 1000 stress_per_atom.txt id type x y z c_peratom[1] c_peratom[2] c_peratom[3] c_peratom[4] c_peratom[5] c_peratom[6]
fix def1 all print 100 "${p1} ${p2}" file Stress_strain.def1.txt screen no
Creating a Movie in OVITO
- Open Ovito.
- Select "File/Open Local File," Choose the LAMMPS dump file you wish to animate, and "Open." For this tutorial select "stress_per_atom.txt".
- You must tell Ovito what each column of data in the dumpfile is. Often, Ovito can do this for you if you click "Auto-assign columns."
- For the c_peratom columns choose stress tensor as the particle property. The 6 stress components are dumped in the order: XX, YY, ZZ, XY, XZ, YZ.
- Use the Modifier List to add the desired modifications to your images and future animation. For this example, the "Color Coding" modifier was used to visualize the stress.
- Under "Color Coding", choose YY as the property. This will animate the stress in the Y-direction (deformation direction).
- Test the animation by using the animation tools below the graphics.
- Click the "Render" tab.
- Select "Complete animation", and under background select "transparent". Select save to file, choose output type (.gif for this example) and choose an output filename in "Output Filename."
- Select the view that you would like to animate and click "Render Active Viewport" above "Render settings."
- Now you should have an animated .gif file for every frame of your simulation.