
×
![Enlarged Image]()
Integrated Computational Materials Engineering (ICME)
How to Calculate Generalized Stacking Fault Energy Curve on (111) Glide Plane, Along [1-10] Glide Direction for Aluminum
Overview
The generalized stacking fault curve represents the energy dependency of a crystal which is rigidly sheared along a plane. Simply it is the Energy-displacement curve as a perfect crystal is sheared.It gives information about a crystals shear properties such as shear strength.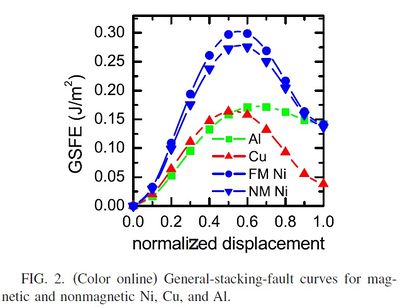
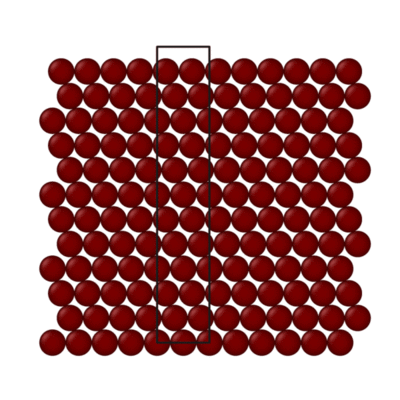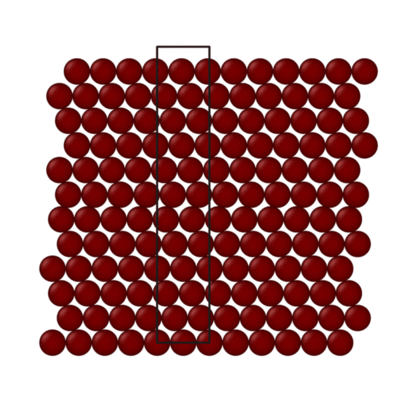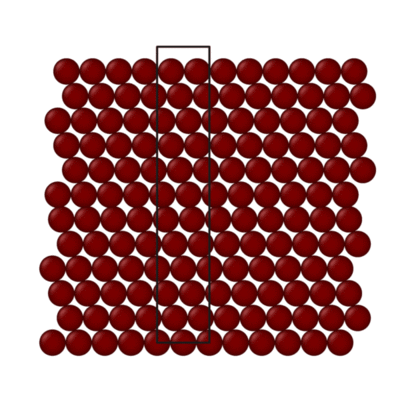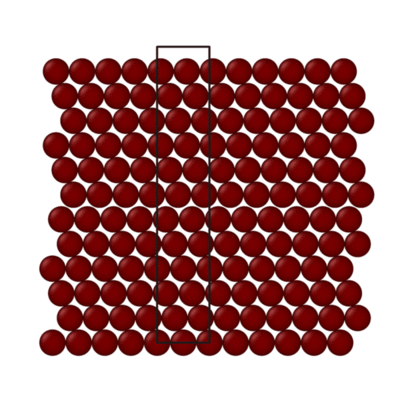
Animation of simulation cell for generalized stacking fault curve for Al (111) surface. Black box indicates simulation cell size.
Preparation
FCC Generalized Stacking Fault Structure Generation Script
Create a directory to run VASP from and save this python script> as a python file in that directory.VASP Input Files
Place the potential file (POTCAR), the input file (INCAR), and K-Points (KPOINTS) in the new VASP directory.Generalized Stacking Fault Energy Curve Generation
Save this BASH Script in the directory and do not give it a file extension.Job/Cluster Submission Files and Executable
In the same directory save the these job submission files. Obtain a VASP executable file from which to run these calculations.Permissions
In a command terminal (LINUX), navigate to the directory containing all the above files. Enter the commandchmod 770 *to open permission editing and execution for the file owner and the usergroup.
Running the Calculation
Use the PBS script provided above to submit the GSFE study to Raptor.To reduce error, first perform a K-Point convergence.