
×
![Enlarged Image]()
Integrated Computational Materials Engineering (ICME)
How to Calculate Basic Calculations to get the Lattice Parameter, Cohesive Energy, and Bulk Modulus for Aluminum
The necessary tools to perform the basic calculations to investigate the ground
state fcc lattice parameter for Al, the cohesive energy and the bulk modulus
determination have been shown in this page.
Please understand that one needs to change the lattice parameter every time to get the different energy values corresponding to different lattice parameter. Rest of the files such as POTCAR, INCAR, KPOINTS remain same, only POSCAR changes. At the end of this page a script file is provided in order to run VASP for several lattice parameters. Also a binary file that can produce the equilibrium lattice parameter, cohesive energy, and bulk modulus, is attached.
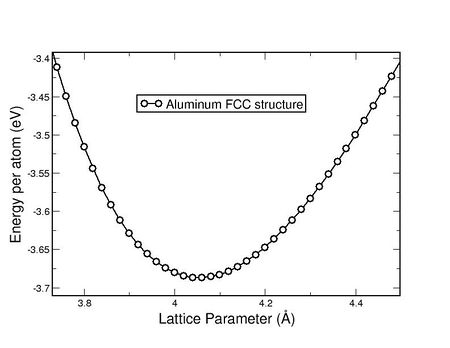 Where the lattice parameter A0 = 4.05725 Angstrom. Bulk Modulus K0 = 751.27
Kbar/10 = 75.12 GPa. Cohesive energy Emin = -3.688 eV.
Where the lattice parameter A0 = 4.05725 Angstrom. Bulk Modulus K0 = 751.27
Kbar/10 = 75.12 GPa. Cohesive energy Emin = -3.688 eV.
Please understand that one needs to change the lattice parameter every time to get the different energy values corresponding to different lattice parameter. Rest of the files such as POTCAR, INCAR, KPOINTS remain same, only POSCAR changes. At the end of this page a script file is provided in order to run VASP for several lattice parameters. Also a binary file that can produce the equilibrium lattice parameter, cohesive energy, and bulk modulus, is attached.
POSCAR
The following code defines the box size corresponding to the near equilibrium fcc lattice structure of one Al atom:Al_FCC a= 4.05 #Header line 4.05 #Scaling parameter to the next three lines of lattice vectors 0.000 0.500 0.500 #Three basis vectors 0.500 0.000 0.500 0.500 0.500 0.000 1 # No of atoms Direct #Could be direct or cartesian 0.0000000000 0.0000000000 0.0000000000 #atom positions
POTCAR
The Potential Card can be downloadable from: /usr/local/vasp/ Depending on which potential you would like to use, go to:- PAW (Projected augmented wave),
- PAW-GGA (Projected augmented wave-generalized gradient approximation),
- PS (Pseudopotential) etc.
KPOINTS
The number of nodal points to calculate the solution; in mesh refinement the greater the number the better the answer but is more costlyMK11x11x2 #header file 0 Monkhorst #Style of Kpoints 11 11 11 #Numbers 0 0 0
INCAR
Input Card to solve the Schrodinger EquationISMEAR = -5 IBRION = 2 ISIF = 3 LWAVE = .FALSE. LCHARG = .FALSE. ENCUT = 250.00
Sample Script #1
To run VASP for several lattice parameters between 3.5 and 4.5 use the shell script below:rm Summary latconst energies evfit.* for a in `seq -w 3.5 0.02 4.5` do echo "a= $a" cat >POSCAR <> Summary done
Sample Script #2
Sample output from Script #1 with different lattice parameters (first column below) and the energy (second column below). The minimum energy is the cohesive energy, which is then used to get the bulk modulus:The "evfit" program basically takes the data for the energy values corresponding to the different lattice parameter as produced by the script shown above in file "SUMMARY". For Al first few lines of that file "SUMMARY" is given below: ......................... 3.920000 -3.643651 3.940000 -3.656043 3.960000 -3.666243 3.980000 -3.674341 4.000000 -3.680354 4.020000 -3.684328 4.040000 -3.686602 4.060000 -3.687003 4.080000 -3.685699 4.100000 -3.682841 4.120000 -3.678444 4.140000 -3.672591 4.160000 -3.665411 4.180000 -3.656935 ................... ................... And produce the minimum of the energy volume curve, as shown below: Equation of state: Murnaghan. CHISQ = 0.1782D-06 A0= 4.0572576 K0= 751.271068 Kbar DK0= 3.8322151 D2K0= 0.0000000 Emin= -3.68763243 3.500 -2.611464 -2.609253 -0.002211 3.520 -2.706994 -2.706268 -0.000726 3.540 -2.796378 -2.796741 0.000363 3.560 -2.880139 -2.881033 0.000894 3.580 -2.958324 -2.959482 0.001158 3.600 -3.031111 -3.032406 0.001295 3.620 -3.099039 -3.100102 0.001063 ............................................... ...............................................
Figure 1. Energy Vs Lattice parameter for fcc Al. Note below the Al properties from this simulations are closely matching with any experimental results.