
Integrated Computational Materials Engineering (ICME)
MnBi
Abstract
We investigated the substitution of Ni atom in MnBi. DFT with GGA functional indicate that substitution energies of these elements are negative. However, DFT+U method calculated the substitution energies to be small positive number for substituted Ni. We also compared the total energies of three different structures using HSE method so there is no Ueff. Our results show that Ni atom go to the regular Mn lattice site in MnBi unit cell equally likely with ferromagnetic and antiferromagnetic spin configuration.
Computational Method
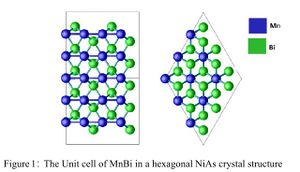
MnBi has a hexagonal NiAs crystal structure that belongs to P63/mmc space group. Fig [1] shows a unit cell of MnBi used in the present work that contains four atoms of two formula units. Magnetism in MnBi arises from Mn atoms occupying 2a site while Bi occupies 2c site.[1] Total energies and forces were calculated using DFT with projected augmented wave (PAW) potentials as implemented in The Vienna Ab initio Simulation Package (VASP). All calculation were spin polarized. A plane wave cutoff of 520 eV was used for pure MnBi and substituted MnBi. Reciprocal space was sampled with a 8x8x6 Monkhorst-Pack mesh. Electron exchange and correlation was treated with GGA as parameterized by the Perdew-Burke-Ernzerhof (PBE) scheme. To describe MnBi and substituted MnBi system correctly, we used GGA+U as well as HSE methods.
Results and Discussion
Pure MnBi
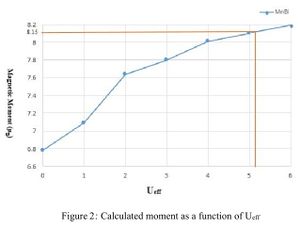
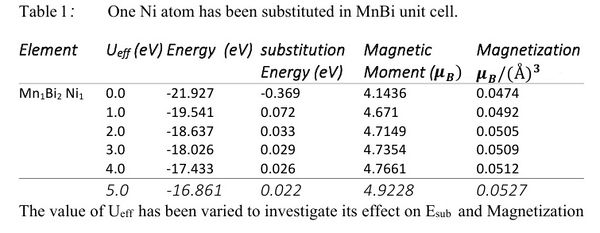
We used DFT + U method to describe strongly correlated d-electrons of Mn atoms in MnBi. We performed a series of total energy calculations for different Ueff values. It has been noticed that the total magnetic moment of the unit cell increase as Ueff of Mn atom increases. To select the best Ueff value for Mn we performed a HSE calculation. Although HSE calculations are computationally much more demanding, it is considered to be more reliable than DFT+U calculations. HSE calculations estimated the total magnetic moment to be 8.153μΒ. As shown in Fig[2] if we set Ueff to be around 5 eV we get around 8.15μΒ. As shown in Fig[2] if we set Ueff to be around 5 eV we get around a = 4.48 Α° and c - 6.058Α° while the experimental lattice constants of pure MnBi are a - 4.256Α° and c = 6.042Α° at 4.2 K.[2] Table [1] shows the total energy of the MnBi unit cell and magnetization for Ueff =0.0 and 5.0 eV.
Ni substituted MnBi
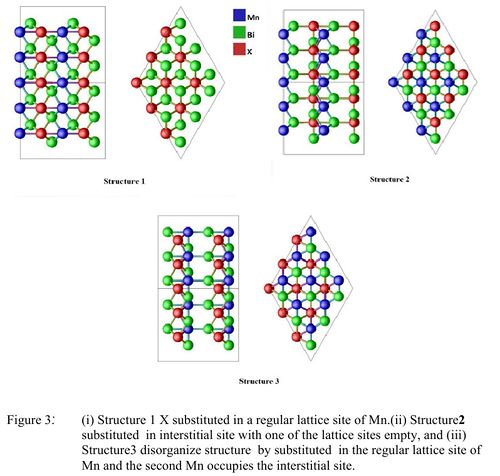
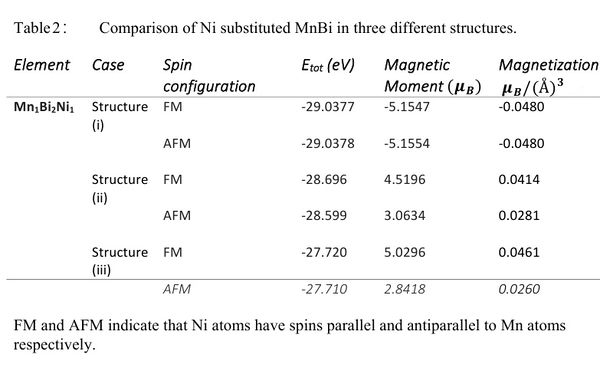
In this case we added one Ni atom and removed one Mn atom from the unit cell of MnBi in Structure[1]. Table [2] shows the effect of Ueff on the substitution energy and magnetization. For Ueff = 0 the substitution energy is negative which indicates that Ni substituted MnBi can be synthesized experimentally. However for other values of Ueff the substitution energy is a small positive number (< 1 eV). Considering the fact we have not investigated the full phase space and we do not know Ueff of Mn and Ni which depends on neighboring atoms, a small positive value of substitution energy does not eliminates the possibility of formation of Ni substituted MnBi. Next we investigate the possibility where substituted Ni atom does not go to the regular Mn site but forms Structure [2] or Structure [3]. Table[3] compares the total energy of three cases. Here we have used HSE method which is considered to be a more accurate method without adjustable parameter Ueff. We see in the Table[3] that the energy of the unit cell in Structure [1] is the lowest energy. This indicates that the substituted atom takes a regular Mn site equally likely with ferromagnetic and antiferromagnetic Spin configuration
Reference
1. Christopher, N., Mishra, S, Singh, N., Singh, S., Gahtori, B., Dhar, A.,
& Awana, V. (2013). Appreciable magnetic moment and energy density in
singlestep normal route synthesized MnBi. Journal Of Superconductivity And
Novel Magnetism, 26(11), 3161-3165
2. [ROB56] Roberts, B. (1956). Neutron diffraction study of the structures and
magnetic properties of manganese bismuthide. Physical Review, 104(3),
607-616