
Integrated Computational Materials Engineering (ICME)
Modified Embedded-Atom Method Interatomic Potentials for the Mg-Al Alloy System
Abstract
We developed modified embedded-atom method (MEAM) interatomic potentials for the Mg-Al alloy system using a first-principles method based on density functional theory (DFT). The materials parameters, such as the cohesive energy, equilibrium atomic volume, and bulk modulus, were used to determine the MEAM parameters. Face-centered cubic, hexagonal close packed, and cubic rock salt structures were used as the reference structures for Al, Mg, and MgAl, respectively. The applicability of these MEAM potentials to atomistic simulations for investigating Mg-Al alloys was demonstrated by performing simulations on Mg and Al atoms in a variety of geometries. These MEAM potentials were used to calculate the adsorption energies of Al and Mg atoms on Al (111) and Mg (0001) surfaces. The formation energies and geometries of various point defects, such as vacancies, interstitial defects, and substitutional defects, were also calculated. We found that the MEAM potentials give a better overall agreement with DFT calculations and experiments when compared against the previously published MEAM potentials. Full Article
Introduction
Magnesium alloys are becoming increasingly important in many technological areas, including aerospace and automotive industries. The usage of magnesium die castings, for example, is increasing in the automotive industry1–3 due to the lower mass densities of magnesium alloys compared with steel and aluminum, higher temperature capabilities and improved crash worthiness over plastics. The primary magnesium alloys for die-casting applications are the magnesium-aluminum alloys such as AM50 and AM60B.4–6 To meet the industrial demand for high-strength lightweight magnesium alloys, it is essential to obtain detailed understanding of the effect of individual alloying elements on the properties of magnesium alloys, especially among the main constituent elements, Mg and Al. The alloying elements can form interstitial or substitutional defects, or can precipitate into small particles creating complex interface structures. The interactions between these alloying elements need to be investigated using atomistic simulation techniques such as molecular dynamics or Monte Carlo simulations. These atomistic simulations require accurate atomic interaction potentials to compute the total energy of the system. First principles calculations certainly can provide the most reliable interatomic potentials. However, realistic simulations of alloy systems often require a number of atoms that renders these methods impractical—they either require too much computer memory or take too long to be completed in a reasonable amount of time. One alternative is to use semi- empirical interaction potentials that can be evaluated efficiently, so that the atomistic approaches that use them can, in certain cases, handle systems with more than a million atoms.
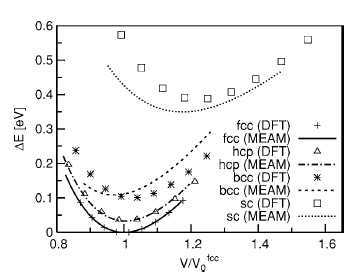
Fig. 1. Atomic energies as a function of the atomic volume for Al atoms in fcc, hcp, bcc, and simple cubic crystal structures. The energies are measured from the equilibrium atomic energy of fcc structure. Volumes are scaled by the equilibrium atomic volume of the fcc structure V0 fcc.
Validation
To test the validity of the MEAM potentials for single elements, each element was put into fcc, hcp, body-centered cubic (bcc), and simple cubic (sc) crystal structures. The atomic energies for several atomic volumes near equilibrium atomic volume were calculated. The results were compared with those of DFT calculations, as shown in Fig. 1. As expected, the curve for the fcc structure produced by the MEAM potential retraces the results of DFT calculations nearly perfectly since fcc was used as the reference structure during the potential construction process. The agreement between the MEAM potential and DFT for the hcp structure is also remarkable. The most important result, however, is the fact that the new MEAM potential correctly identified fcc as the most stable structure for Al. Furthermore, the sequence of the structures is correctly predicted in the order of stability by the Al MEAM potential. The relative cohesive energies, with respect to the one for the fcc structure, are also in good agreement with the DFT calculations, although the result for the simple cubic structure is slightly underestimated. The relative equilibrium atomic volumes, with respect to the one for the fcc structure, are also well reproduced. We point out that the equilibrium atomic volume for fcc Al, obtained by the MEAM potential (16.61 Å^3), is slightly different from the one predicted by DFT (15.76 Å^3). This is due to the fact that the MEAM parameters are fitted to reproduce the experimental volume, while DFT within LDA tends to underestimate the equilibrium lattice constants by roughly 1%. Figure 2 shows the atomic energy plot for Mg atoms in different crystal structures compared with the results of the DFT calculations. The hcp structure was used as the reference structure for the Mg MEAM potential, and the DFT data points for this structure are accurately reproduced. The sequence of the structures is again predicted correctly in the order of stability by the Mg MEAM potential. The relative atomic energies, with respect to the one for the hcp structure, are also in good agreement with the DFT calculations. Note that the scale of the vertical axis of Fig. 2 is six times larger than that of Fig. 1, and that the largest error in relative atomic energies is in the order of 0.01 eV. Similar to the Al potential, equilibrium atomic volume for hcp Mg in the MEAM is set to the experimental value of 23.16 Å^3, while DFT predicts a smaller value of 21.54 Å^3. Both the MEAM and the DFT methods prefer a c/a ratio close to 0.994 of the ideal c/a ratio.
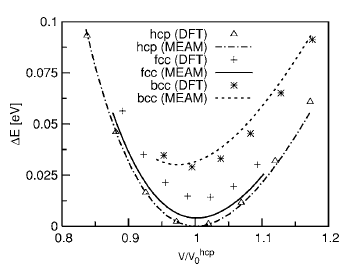
Fig. 2. Atomic energies of Mg as a function of the atomic volume in fcc, hcp, and bcc cubic crystal structures. The energies are measured from the equilibrium atomic energy of the hcp structure. Volumes are scaled by the equilibrium atomic volume of the hcp structure V0 hcp.
Conclusion
In this study we developed a set of MEAM potentials for Al, Mg, and their alloy systems using first-principles calculations based on DFT. The validity and transferability of these MEAM potentials were tested rigorously by calculating physical properties of the Mg-Al alloy systems in many different atomic arrangements such as bulk, surface, and point defect structures. These MEAM potentials show a significant improvement over the previously published potentials, especially for the surface formation, stacking faults, and point defect calculations. The Mg-Al alloy potentials, however, failed to predict the stability of the Mg17Al12 alloy intermetallic phase, suggesting the need for further improvements.